| 导 读
2019年8月5日,百济神州(北京)有限公司的化学首席研发总监王志伟博士等在J. Med. Chem(IF: 6.259) 杂志上,发表题为“Discovery ofZanubrutinib (BGB-3111), a Novel, Potent and Selective Covalent Inhibitor ofBruton’s Tyrosine Kinase”的论文。研究人员通过体外药效学、激酶选择性、药代动力学和体内药效实验对化合物进行了系统的筛选,发现了一种高效、高选择性的不可逆BTK抑制剂BGB-3111(31a,Zanubrutinib)。
| 内 容
布鲁顿的酪氨酸激酶(BTK)属于胞质酪氨酸激酶的TEC家族,是人类第二大非受体激酶家族。它在造血系统的所有细胞系中均可表达,除T细胞外,还存在于骨髓、脾脏和淋巴结组织中。编码BTK基因的失活突变可导致人类X-连锁无丙种球蛋白血症(XLA)和小鼠的X-连锁免疫缺陷(XID)。这些疾病的特点表现在B细胞发育和功能上出现明显缺陷,提示BTK对B细胞发育和功能有重要作用。此外,BTK在B细胞中的激活后会引发自身激活的血浆细胞的积累。BTK可在BCR信号通路中被上游SRC家族激酶激活,一旦被激活,BTK反过来磷酸化磷脂酶Cγ(plcγ),导致Ca2+活化和同时激活NF-κB和MAP激酶途径。这些近端信号事件促进了BTK参与增殖和存活的基因表达。除了作为BCR下游的重要调节作用外,BTK在FC受体(FCR)信号传导中也起着关键作用。通过FcγR相关受体的信号传导也促进巨噬细胞等产生BTK依赖性促炎性细胞因子。由于BTK位于BCR和FCR信号通路中的近端位置,所以它一直是一个重要的靶点。同时,BTK的异常激活在B细胞淋巴瘤发病机制中起着关键作用,提示BTK的抑制在治疗血液恶性肿瘤中有重要贡献。
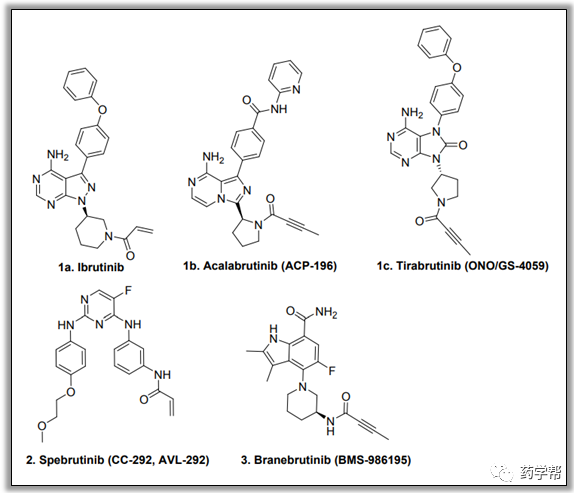
图1:临床上已知的不可逆BTK抑制剂的结构(图片来源于JMC期刊)
第一个临床有效的共价BTK抑制剂Ibrutinib(图1)被美国FDA批准用于治疗慢性淋巴细胞白血病(CLL)、套细胞淋巴瘤(MCL)、巨球蛋白血症(WM)和慢性移植物抗宿主病(cGVHD)。最近已经开发并正在进行临床试验的其他几个共价抑制剂(图1),包括Acalabrutinib(ACP-196),Tirabrutinib(ONO/GS-4059), Spebrutinib(CC-292,AVL 292)和Branebrutinib(BMS-986195)。Acalabrutinib在2017年被美国FDA批准用于治疗MCL。尽管Ibrutinib有很好的疗效和一般耐受性,但有报道称其有出血、皮疹和腹泻等不良事件。这些Ibrutinib相关副作用被认为部分与靶外激酶的抑制有关,如野生型(WT)EGFR、TEC、JAK3和SRC家族成员(如SRC、Lyn、Fyn和LCK)等。众所周知,靶向WT-EGFR可诱导显著的皮肤毒性和胃泌素。由于表皮生长因子受体信号级联与皮肤和胃肠系统生物学有关,因此会产生不良影响。BTK和TEC都属于TEC家族激酶。血小板表达的BTK和TEC,作为糖蛋白VI(GPVI)信号的下游。TEC补偿了小鼠血小板中GPVI下游信号中缺乏BTK的情况。Ibrutinib抑制TEC激酶干扰血小板聚集,可能有助于观察出血。最近的研究表明,通过比较不同的BTK抑制剂在胶原诱导的血小板活化中的活性,BTK抑制是抑制血小板聚集的主要驱动因素。SRC家族激酶如SRC、Lyn、Fyn和LCK也被报告引起与出血风险增加相关的止血功能障碍。尽管Ibrutinib的临床抗肿瘤疗效是否与靶外效应有关尚不确定,但发现靶外抑制剂更具选择性是可取的。
近日,百济神州(北京)有限公司的化学首席研发总监王志伟博士等通过体外药效实验、选择性实验、药代动力学(PK)和体内药效学实验对化合物进行系统的筛选,发现了一种高效、高选择性的不可逆BTK抑制剂BGB-3111(31a,Zanubrutinib),它对BTK具有较强的活性,对其他TEC、EGFR和SRC家族激酶具有良好的选择性,对小鼠具有理想的ADME性质和良好的体内药效。
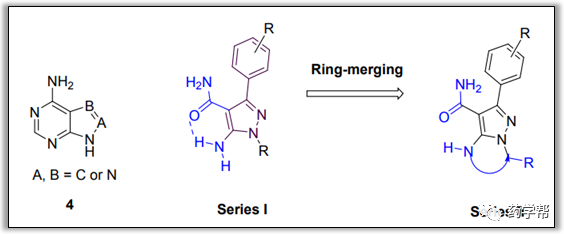
图2:新系列化合物的设计(图片来源于JMC期刊)
众所周知,吡咯嘧啶、吡唑嘧啶和嘌呤片段(图2)常用于激酶抑制剂的设计。通过分子内氢键模拟嘧啶酮环也是药物设计中常用的一种有吸引力的策略。如图2所示,前期首先合成了一个伪嘧啶酮I系列,这类化合物显示出良好的BTK活性。本文作者通过环合并方法设计了一种新的II系列,发现该系列化合物对其他激酶具有高的BTK活性和高的选择性。
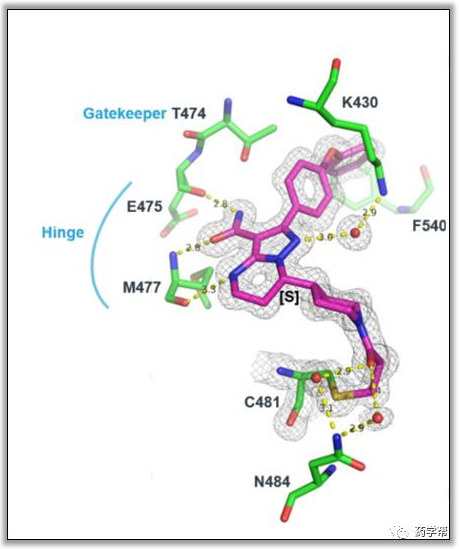
图3: 31a和cys481残基作用图 (PDB ID:6j6m) (图片来源于JMC期刊)
为了考察31a的激酶选择性,研究人员测试了化合物在1μm时对342种人类激酶的抑制活性。结果显示31a对329种人类激酶的抑制率低于70%,对包括BTK在内的13种激酶的抑制率高于70%。因此进一步测试了包括MEK2在内的这13种激酶对31a的IC50。其中10种在类似位置具有类似半胱氨酸残基的激酶抑制实验中,31a对ITK的选择性为187倍,对JAK3的选择性为1933倍,对Her2的选择性为1800倍,这可能归因于化合物固有结构的选择性和这些激酶的半胱氨酸对共价结合的微环境影响。此外,31a在SRC家族激酶如SRC(29%抑制)、Lyn(35%抑制)、Fyn(50%抑制)和LCK(73%抑制,IC50=187nM)中表现出低抑制作用,据报道这些激酶在血小板活化中起关键作用,与出血风险增加相关。对比Ibrutinib,31a对ITK、JAK3、HER2、FGR、CK、CSK、Fyn、HCK、Lyn、SRC、Yes和FRK的激酶活性有更高的IC50值或无抑制作用。
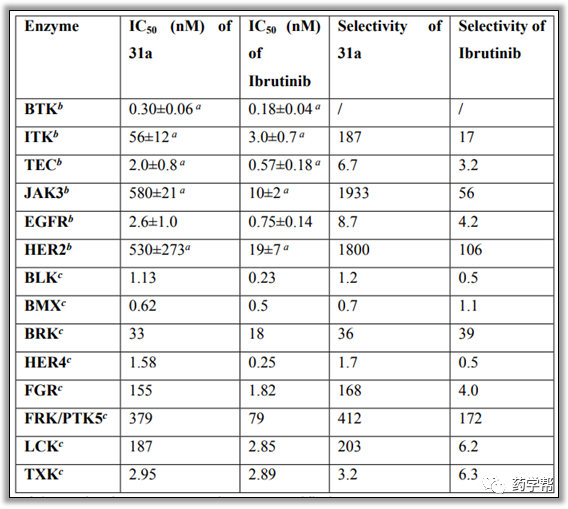
图4: 化合物31a和Ibrutinib的生化激酶选择性(图片来源于JMC期刊)
总结:研究人员采用构效关系驱动的药物设计策略,并通过使用体外药效、选择性、药代动力学和体内药效学来筛选化合物。从一系列拟嘧啶酮类化合物出发,设计合成了两个系列(三环和双环)的新化合物。其中31a是一种高效、特异性高、不可逆的BTK抑制剂。它对其他TEC、EGFR和SRC家族激酶有很强的选择性,表现出优良的ADME性质和小鼠体内药效活性,以及良好的OCI-LY10 DLBCL异种移植模型的疗效。化合物31a目前正在进行临床试验。
附录:合成路线
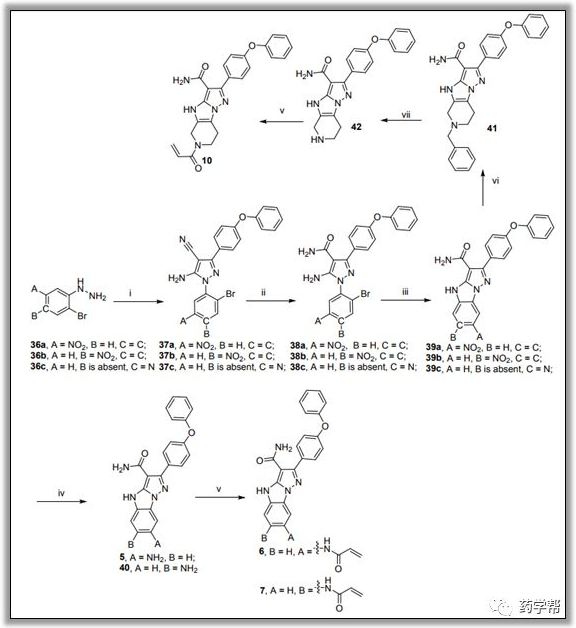
图5. 化合物5,6,7和10的合成通法(图片来源于JMC期刊). 合成条件:(i)EtOH, 2-(methoxy(4-phenoxyphenyl)methylene)malononitrile, 70℃; (ii)phosphorous acid (85% in H2O), 100℃;(iii) DMF, CuI, N1,N2-dimethylethane-1,2-diamine, K3PO4, 60℃; (iv)HOAc, Zn, rt; (v) DCM, acryloyl chloride, TEA, 0℃ tort; (vi) THF, benzyl bromide, 65℃, thenMeOH, NaBH4, rt; (vii) MeOH, Pd/C (10%), H2 (1 atm), rt.
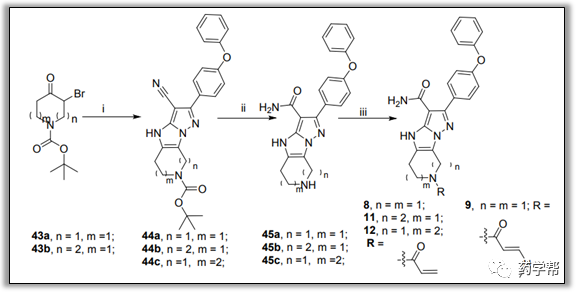
图6. 化合物8,9,11和12的合成通法(图片来源于JMC期刊).(i)5-amino-3-(4-phenoxyphenyl)-1H-pyrazole-4-carbonitrile, DMF, K2CO3,80 ℃; (ii)H3PO4 (85% in H2O), 100 ℃; (iii)DCM, acryloyl chloride, TEA, 0 ℃ to rtor 4- (dimethylamino)but-2-enoic acid hydrochloride, HATU, TEA, DCM, rt.
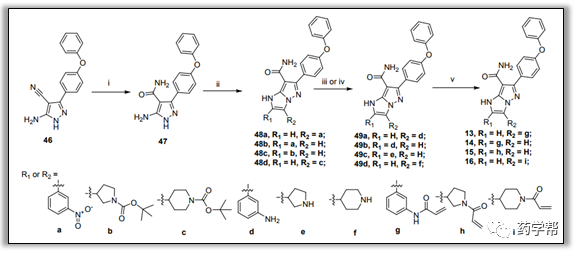
图7. 化合物13-16的合成通法(图片来源于JMC期刊).(i) H3PO4(85% in water), 120℃; (ii)EtOH, haloketone, 80℃;(iii) MeOH/DCM, Pd/C (10%), H2 (balloon), rt; (iv) HCl, MeOH, rt;(v) DCM, TEA, acryloyl chloride, rt.
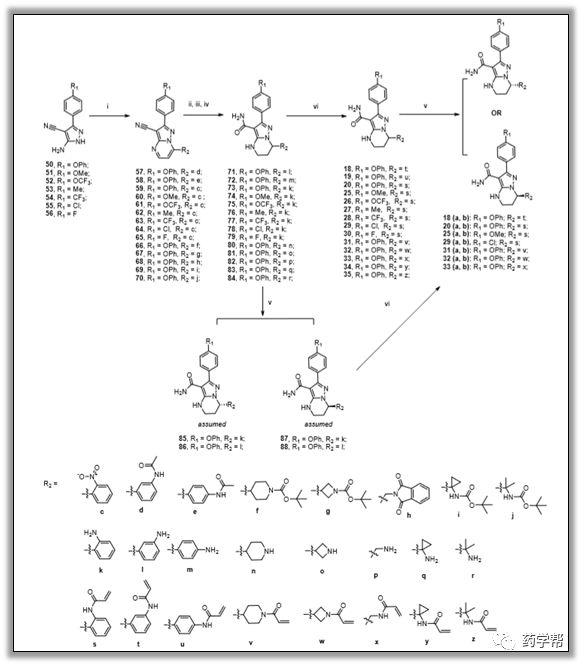
图8. 化合物18-20, 25-35的合成通法(图片来源于JMC期刊). (i)3-dimethylamino-1-(aryl, heteroaryl or alkyl)-2-propen-1-one; acetic acid,reflux; (ii) EtOH or MeOH, HCl (con.), 75℃ or 60℃; orhydrazine hydrate, MeOH/dioxane; 60℃; orPd/C (10%), MeOH/DCM, rt, H2. (iii) EtOH, NaBH4, 60℃; (iv)DMSO/EtOH (v: v = 1:1), 5N NaOH, H2O2 (30%), 60℃; (v)chiral separation; (vi) DCM, pyridine or Et3N, acryloyl chloride,rt.
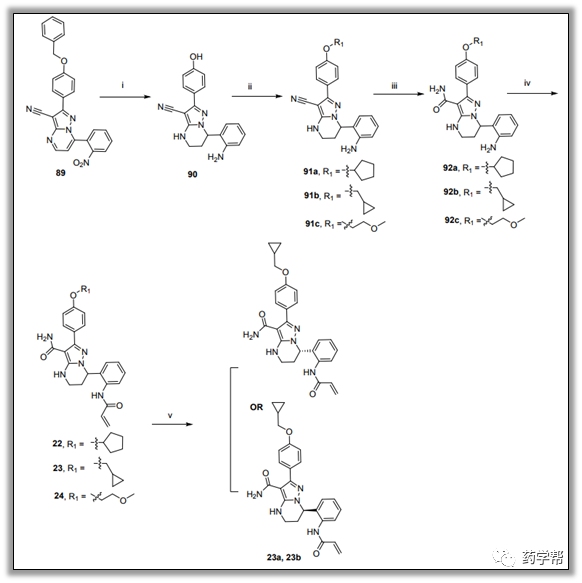
图9. 化合物22-24的合成通法(图片来源于JMC期刊). (i)MeOH/DCM, Pd/C (10%), H2 (3 atm), rt; (ii) acetone, R1Br,K2CO3, rt or reflux; (iii) DMSO/EtOH (v: v = 1:1), NaOH(1N), H2O2 (30%), 60 ℃; (iv)DCM, Et3N, acryloyl chloride, rt; (v) chiral separation.