| 导 读
2019年7月12日, 清华大学饶燏课题组在Journal of Medicinal Chemistry (IF: 6.259) 杂志上,发表题为“Discovery of 4‑Aminoquinoline-3-carboxamide Derivatives as Potent Reversible Bruton’s Tyrosine Kinase Inhibitors for theTreatment of Rheumatoid Arthritis”的研究论文,研究人员利用structure-hopping strategy发现了一系列新的强效、可逆BTK抑制剂——4-氨基喹啉-3-羧酰胺衍生物,用于治疗类风湿类关节炎。该系列衍生物与以往的噌啉类骨架化合物相比较,明显改善了药物的性质,尤其是水溶性。其中,代表性的化合物25不仅对BTKWT ( IC50 = 5.3 nM) and BTKC481S( IC50 = 39 nM)具有较强的抑制作用,而且在啮齿类动物胶原诱导的关节炎模型中,化合物25,在不减轻动物体重的情况下,能有效地减少爪子肿胀情况。通过对药效、药物性质、稳定性和非共价抑制模式考察,该类化合物中具有代表性的抑制剂有望成为治疗广泛的自身免疫性疾病的药物。
| 内 容
类风湿类关节炎(RA)是一种多因素自身免疫性疾病。其特征是手、足小关节的多关节、对称性、侵袭性关节炎症,经常伴有关节外器官受累及血清类风湿因子阳性,可以导致关节畸形及功能丧失。
图片来源于互联网
目前,RA的治疗药物包括非甾体类抗炎药物(NSAIDs)、疾病修饰类抗风湿药物(DMARDs)、TNF - alpha抑制剂、IL-6抑制剂、T细胞活化抑制剂、B细胞耗竭剂、免疫抑制剂等等。2012年,托法替尼(JAK1/3抑制剂)成为第一个获得FDA批准的治疗自身免疫性疾病的激酶抑制剂。通过依法替尼的启发,促使科研工作者们致力于寻找其他与RA相关的激酶靶点。众所周知,BTK在B淋巴细胞的分化、活化、增殖和存活中发挥着重要作用。啮齿动物关节炎模型的研究为托法替尼双重作用机制提供了证据:(i)抑制BCR依赖的B淋巴细胞活化和自身抗体分泌;(ii)抑制髓细胞依赖的炎性细胞因子的产生。
最近,一些研究结果表明,抑制酪氨酸激酶BTK的活性可以为自身免疫性疾病,如RA和系统性红斑狼疮(SLE),提供一个有前景的治疗方案。
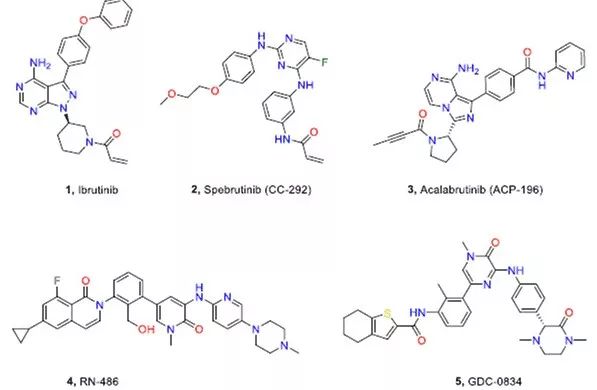
图1 常见的BTK抑制剂 (图片来源于JMC期刊)
近十多年来,几种BTK抑制剂已被发现用于血液恶性肿瘤和慢性炎症疾病的临床治疗。其中,最强效的BTK抑制剂依鲁替尼(Ibrutinib)(图1,1)已被批准用于B细胞恶性肿瘤和慢性淋巴细胞白血病。spebrutinib (CC - 292 图1,2)和acalabrutinib (ACP-196 图1,3)目前也参与了RA的临床开发。但是这些通过共价键发挥不可逆BTK抑制剂存在很多安全风险。如:迈克尔受体的共价键不可逆抑制同时引起了对不相关靶点的抑制。因此,开发可逆的BTK抑制剂对降低RA有效治疗的风险至关重要。
然而,不乐观的是,可逆性BTK抑制剂的开发相对比较滞后,到目前为止,还没有可逆抑制剂被批准用于临床应用。众所周知的非共价BTK抑制剂,RN-486(图1,4)和GDC- 0834(图1,5),也都存在稳定性不好和药代动力学(PK)差的特征。鉴于BTK治疗自身免疫性疾病的发展趋势,开发具有良好药物性质的新型可逆抑制剂迫在眉睫。
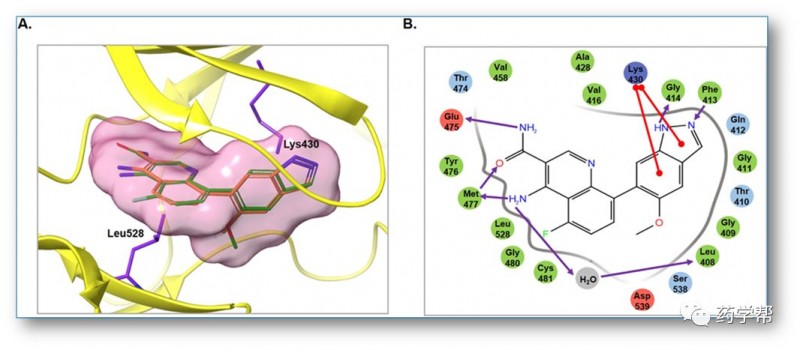
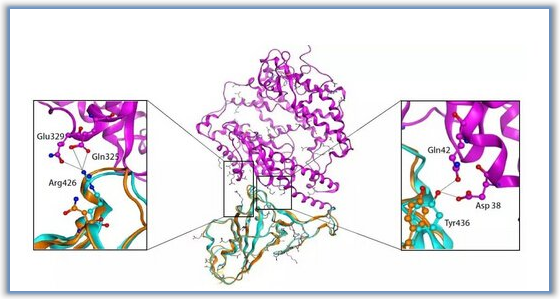
图2 4-氨基喹啉-3-羧酰胺衍生物对接研究(图片来源于JMC期刊)。(A) BTK激酶区域内化合物7(橙色)和25(绿色)的假定结合模式(黄色卡通)。所选的残基用棒表示。(B)受体与化合物25相互作用的简图。深蓝色球(正电荷相互作用);青色与红球(极性相互作用);绿球(疏水相互作用);紫色箭头(H-bond);红线(π阳离子相互作用)。
饶燏课题组采用structure-hopping strategy,发现了4-氨基喹啉- 3-羧基衍生物作为BTK的有效抑制剂。去除不显著的氮,加入甲氧基和氟基取代物,在不影响BTK活性的前提下,显著提高了BTK的水溶性。最有效的抑制剂表现出对BTK和细胞Tyr223自磷酸化的强抑制作用。
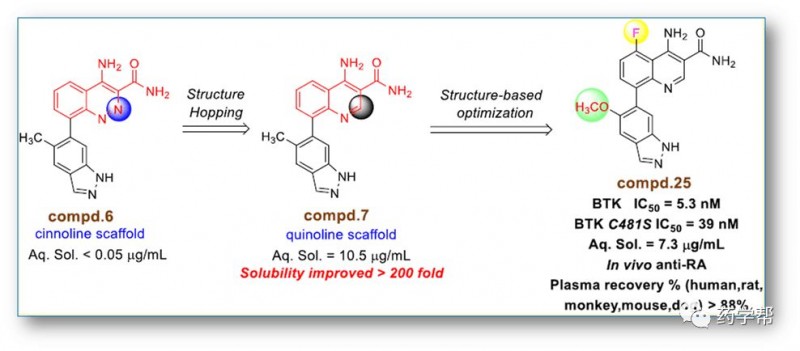
图3 图片来源于JMC期刊
令人欣喜的是,化合物25在BTK活性位点的建模表明,饶燏课题组的可逆抑制剂的结合位姿与依鲁替尼的结合位姿是呈正交的。这样以来就导致化合物25对BTKC481S蛋白的IC50值为39 nM,与Ibrutinib相比,化合物25对EGFR、TEC和ITK 具有更好的激酶选择性。同时,在啮齿动物胶原诱导的关节炎(CIA)模型试验中,化合物25在不减轻体重的情况下有效地减少了爪子肿胀,证明了抑制剂的有效性及耐受性。
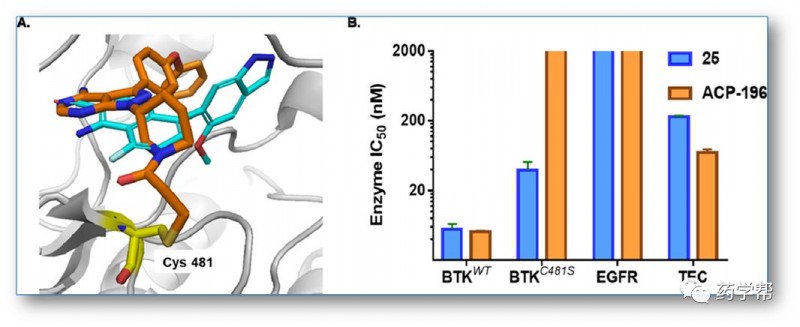
图4 化合物25与BTK的激酶结构域结合的方式不同于现有的ATP模拟共价抑制剂(图片来源于JMC期刊)。(A) Ibrutinib的共晶结构叠加(橙色,PDB代码:5P9J)和25的对接姿态(蓝色)。Ibrutinib与Cys481残基形成共价键,占据守门残基Thr474后的疏水口袋,不同于化合物25。(B)化合物25与ACP-196对BTK、BTKC481S、EGFR和TEC激酶的 IC50值。
总结:清华大学饶燏课题组报道了一系列新型的4-氨基喹啉-3-羧胺类化合物作为强效可逆BTK抑制剂,明显改善了类药物性能,尤其是水溶性。化合物25对BTK具有较强的抑制作用,酶促IC50值5.3nM,细胞自磷酸化抑制EC50值42.7nM。正如对接研究所支持的,氟和甲氧基取代物的加入通过引入额外的相互作用(Leu528与Lys430π−阳离子疏水作用),导致抑制剂效力增加了10倍以上并极可能调整了束缚构象。
在BTK的激酶域的结合方面,饶燏课题组的抑制剂对相关的TEC和SRC家族激酶具有更好的选择性。另外,在啮齿类动物CIA模型中,代表性化合物25在不影响小鼠食欲的情况下,有效地减少了爪子肿胀,这意味着饶燏课题组的抑制剂在本研究中是有效的,耐受性良好。此外,血浆恢复试验和大鼠药代动力学研究表明,该化合物具有良好的药代动力学特性。
综上所述,化合物25被认为是一种新型的、强效的可逆抑制剂,极有可能作为一种治疗广泛的自身免疫性疾病RAs的口服药物。
附录:化合物合成路线
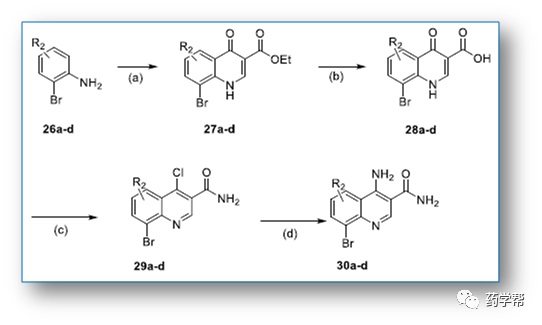
Scheme 1.喹啉中间体的合成(图片来于JMC期刊)
Reagentsand conditions: (a)(i)diethyl ethoxymethylenemalonate,100°C, 2h, (ii) Dowtherm A, 250°C,4−12h, 40−60% (b)LiOH,THF/EtOH/H2O = 4:4:2, 55°C, 4−8h, 75−82%; (c)(i) SOCl2,DMF, 70°C, 3−6h, (ii)NH3·H2O, DCM, 0°C-rt, 1h, 40−60%;(d)7 M NH3 in MeOH, 80°C, overnight, 50−55%.
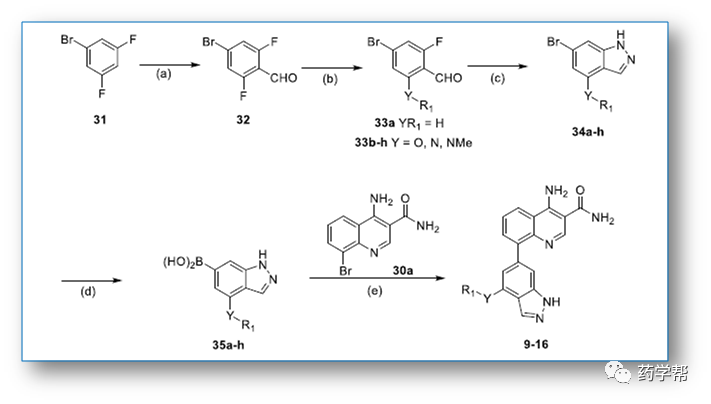
Scheme 2. 化合物9-16a的合成路线(图片来于JMC期刊)
Reagents and conditions: (a)diisopropylamine, n-butyllithium, DMF,anhydrous THF, −78°C-rt, 3h, 50%; (b)R1YH, K2CO3, DMA, 100°C,3−8h, 35−61%; (c) N2H4·H2O/DME 1:1, 170−200°C, 24−48xh, 20−62%; (d)t-buLi, tributyl borate, anhydrous THF, −78°C-rt, overnight,21−62%; (e)Pd(dppf)Cl2, 2M Na2CO3, 1,4-dioxane,microwave, 140°C, 1−2h, 23−84%
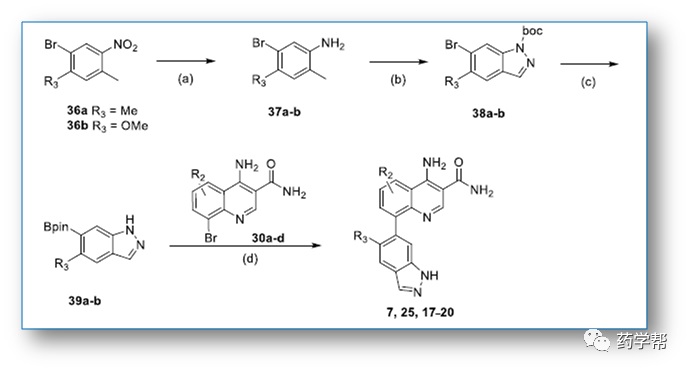
Scheme 3. Synthetic Route of Title Compounds 7, 25, and 17−20a
Reagentsand conditions: (a)Fe, NH4Cl, EtOH/H2O 3:1, 80 °C,1 h, 80−90%; (b)(i) 50% HBF4, NaNO2,H2O, 0°C, 3h; (ii)AcOK, 18-crown-6, CHCl3,rt, 3 h; (iii)Boc2O, DMAP, Et3N, MeCN, rt, 1 h, 25−35%; (c)(i)Pd(dppf)Cl2, B2pin2,KOAc, 1,4-dioxane,90 °C, overnight; (ii)TFA/DCM 1:1, rt, 2 h, 55−60%; (d)Pd(dppf)Cl2, 2 M Na2CO3,1,4-dioxane, 140°C, microwave, 1−2 h, 44−65%.
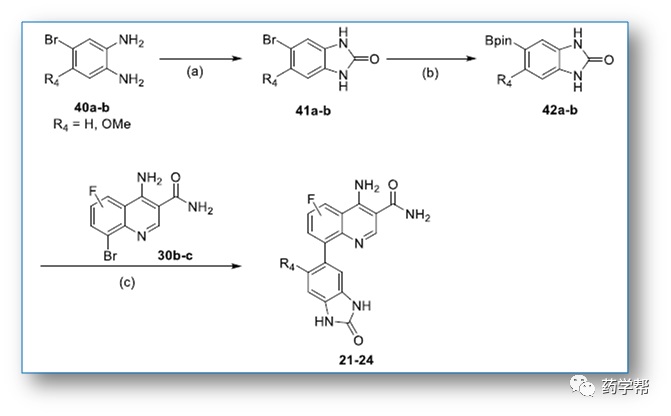
Scheme 4. 21−24的合成路线
Reagents and conditions:(a)N,N′-carbonyldiimidazole,rt,overnight,75−78%;(b)Pd(dppf)Cl2, B2pin2, KOAc,1,4-dioxane, 90°C, 6h, 80−83%;(c)Pd(dppf)Cl2,2M Na2CO3, 1,4-dioxane,microwave, 140°C,2h, 43−51%.
✔饶燏教授简介

饶燏,研究员、博士生导师(图片来源于清华大学官网)
【个人简介】
饶燏副教授,研究员、博士生导师、教育部青年长江学者,国家优秀青年科学基金,教育部新世纪优秀人才和树兰医学青年奖获得者。研究方向:发展小分子诱导蛋白质降解技术(PROTACs)进行靶向蛋白降解及相关药物研究;依据蛋白质三维结构进行抗感染疾病, 抗肿瘤小分子化合物的设计,合成与开发;具有高生物化性小分子的化学合成及作为分子探针应用于化学生物学研究。
【社会兼职】
担任美国化学学会杂志ACS Medicinal Chemistry Letter编委,中国化学快报Chinese Chemical Letters青年编委,Chinese Journalof Medicinal Chemistry《中国药物化学杂志》编委。
【获奖荣誉】
饶燏副教授先后获得树兰医学青年奖、教育部青年长江学者、国家优秀青年科学基金、拜尔研究员奖、清华大学“221”基础研究青年人才、教育部新世纪优秀人才、拜尔青年研究员奖、罗氏优秀研究奖等。
【研究成果】
饶燏教授长期致力于化学合成和药物化学有机结合的研究,发展和建立了一系列新颖高效的C-H 官能团化的合成方法学(如C-H 卤化,C-H 羟基化)和具有一定普适性的药物片段修饰策略,增加了在药物分子设计和合成中的多样性优化手段,为系统深入研究其生物功能奠定了基础。另外,其课题组近年来围绕抗感染(如丙肝,疟疾,登革热等),抗肿瘤(如肺癌,结直肠癌等)疾病的药物开发开展了一系列具有原创性和系统性的研究工作,并申请了多项药物专利。并在Nature、Cell等国际期刊上发表近10篇高水平文章。