| 导 读
2019年5月15日,北京生命科学研究所李超课题组在《Journal of the American Chemical Society》发表题为“Tandem Decarboxylative Cyclization/ Alkenylation Strategy for Total Syntheses of (+)-Longirabdiol, (-)-Longirabdolactone, and (-)-Effusin”的论文。该研究发现了一种金属铜催化的脱羧/环化/烯化串联反应,并成功地应用于C19位被氧化的贝壳杉烷型二萜的首次全合成。
| 内 容
贝壳杉烷型二萜是一类结构非常复杂且具有重要生物活性的天然产物。目前,已有近千个此类化合物从植物中分离得到。在化学结构上,此类天然产物往往含有复杂的环系骨架结构(包括并环,螺环和桥环)和高度的氧化态,是有机合成界关注和研究的重点。在生物活性上,此类天然产物往往展现出抗菌,抗肿瘤和抗病毒等重要活性。其母体植物(香茶菜属植物)是一类常见的抗炎症中药,如冬凌草。具有高度氧化态的此类天然产物是有机合成的难点之一,尤其是C19位被氧化的螺环内酯类贝壳杉烷型二萜,因为C19位的选择性氧化会引起C4产生一个季碳中心,所以目前还没有成功的全合成报道。
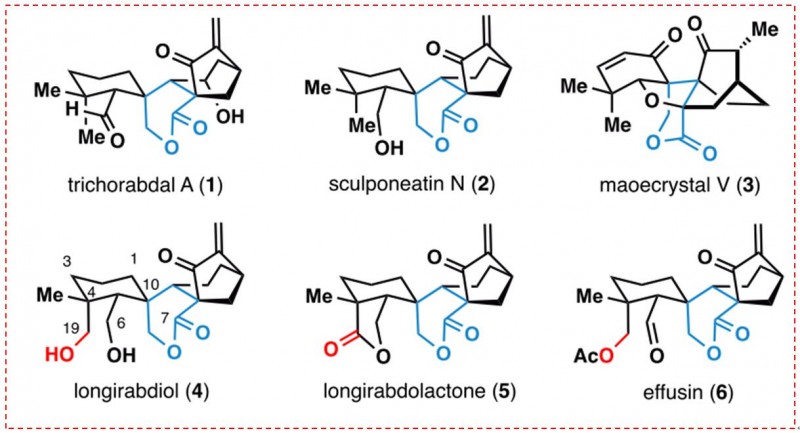
图1:螺环内酯类贝壳杉烷型二萜(图片来源于JACS期刊)
在这项研究中,研究人员首先根据这类复杂天然产物的结构特征,设计并实现了一种高效的串联环化/烯化反应。该关键反应首次揭示了廉价金属铜催化的脱羧/环化/烯化串联偶联反应:从简单易得且稳定地原料出发,一步获得具有较高复杂度且具有多个手性中心的关键顺式内酯类中间体。在这过程中,研究人员通过筛选发现了一个关键添加剂(2-溴丙酸)可以很好地提高整个串联反应的效率,从而高效地构建了具有环张力的五元内酯(环张力的产生源自于C4位的季碳中心)。进一步地底物适用范围拓展发现,该合成方法具有很好的实用性,这也为合成具有更高氧化态的此类天然产物提供了很好的合成方法。
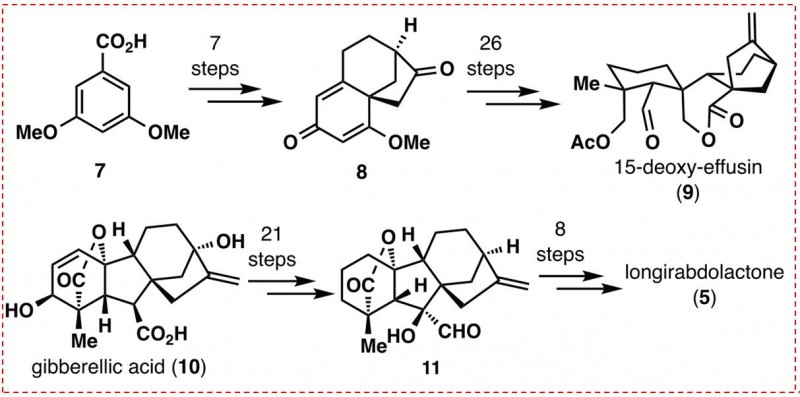
图2:过去的报道(图片来源于JACS期刊)
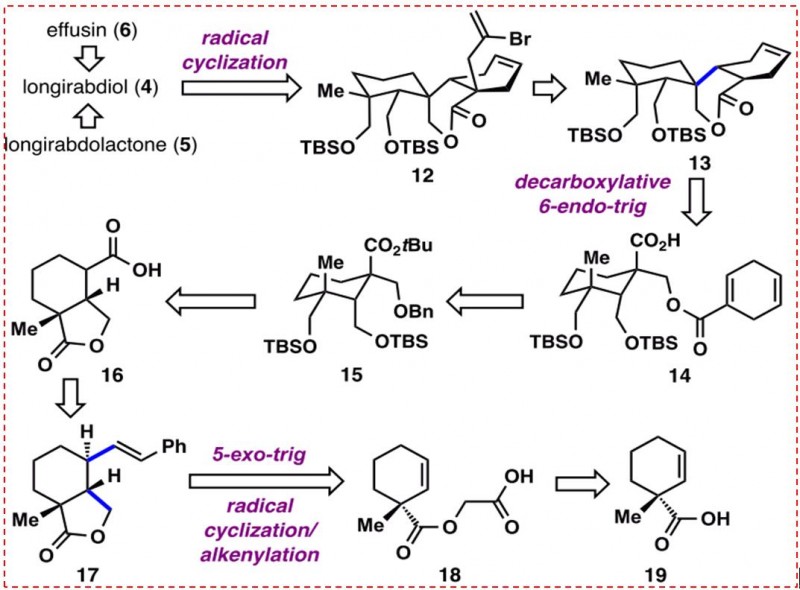
图3:拟合成分析(图片来源于JACS期刊)
研究者设想通过氧化,可以从长链二醇(4)中分离得到长链杜拉酮(5)和(6)。通过经典的乙烯基自由基环化,可以获得4的双环辛烷环9(9结构见图2)。12中的乙烯基侧链可以通过13上的烯丙基化来安装。由羧酸14经脱羧基6-内三角自由基环化得到含有γ-二取代的δ-缬内酯的化合物13。化合物14可以很容易地由苄基醚15合成。15的羧酸酯可进一步与酸16断开,16的羧酸可通过17中烯烃部分的氧化裂解而得到。关键中间体17的合成,作者采用了串联脱羧环化/烯化策略(图4),以高的立体选择性地从羧酸18合成了γ-内酯17。
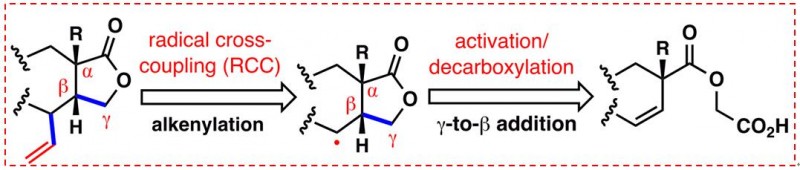
图4:串联脱羧环化/烯化策略(图片来源于JACS期刊)
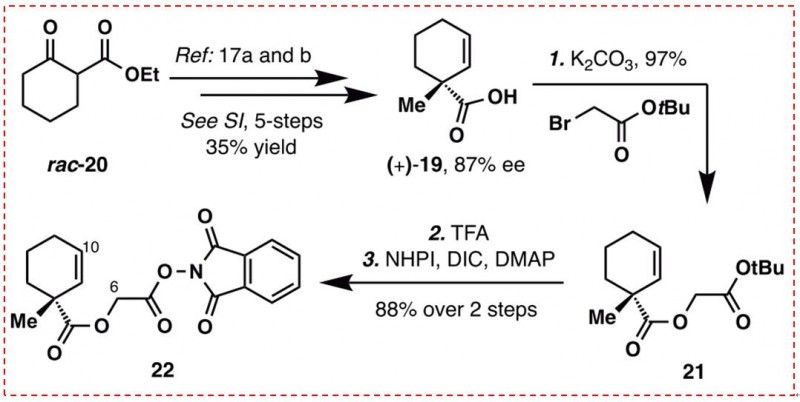
图5:氧化还原活性酯22的合成(图片来源于JACS期刊)
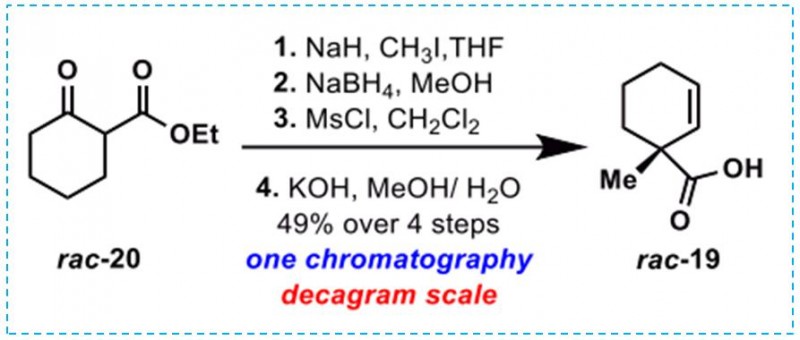
附图:(+)-(19)的合成(图片来源于JACS期刊)
化合物(+)-(19)是通过rac-20(市售)经过5步反应以87%的ee值和35%的总收率制备而得。(+)-(19)再次与溴乙酸叔丁酯缩合,提供了一种可以用N-羟基邻苯二甲酰亚胺(NHPI)活化的羧酸酯,以高的产率得到氧化还原活性酯22,合成路线如图5所示。
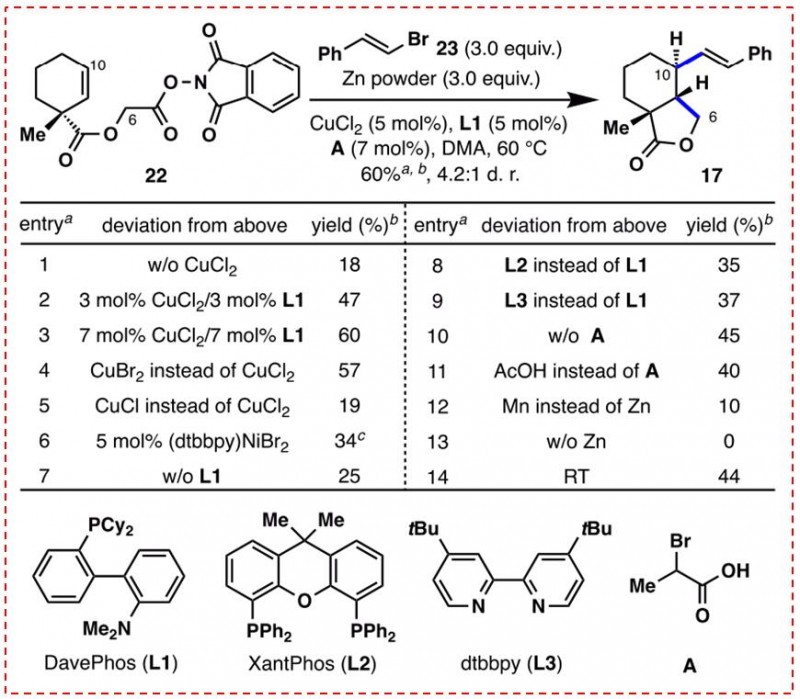
图6:自由基环化/烯化反应的优化(图片来源于JACS期刊)
设想22可以通过单电子还原脱羧,提供一个可以参与5位-外三环化反应的中间体,由此产生的二级自由基可参与连续的烯化反应。在最初的研究中使用了乙烯基锌试剂,这与Baran组在脱羧烯化方面的开创性研究非常相似。然而,22与乙烯基锌试剂使用各种镍或铁配体配合物的反应导致了C6位的直接烯化。这可能归因于铁或镍催化的脱碳反应的反应速率太快。研究者更换为Reisman和 Weix组开发的RAE和SP2-卤化物之间的还原交叉耦合反应方法。令人欣慰地,22和β-溴苯乙烯暴露于(DTBBPY)NiBr2和锌粉中,以34%的产率提供所需产物17,作为3.2:1非对映体混合物(图6)。通过进一步优化反应条件,将反应收率提高至60%(4.2:1非对映体混合物)。
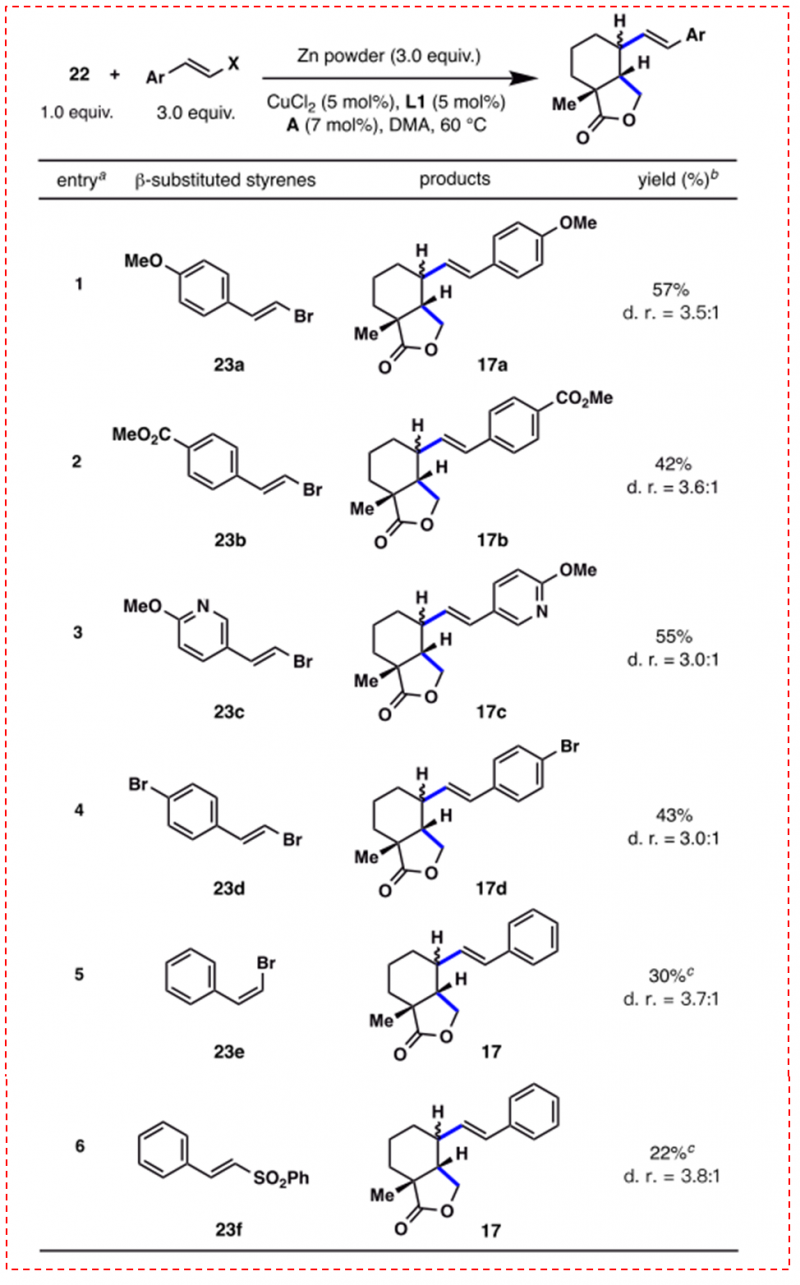
图7:β-取代苯乙烯的应用范围(图片来源于JACS期刊)
获得最佳优化条件后,为了进一步了解其潜在的反应机理,研究者应用此串联环化/烯化反应,进一步探索了一系列具有代表性的β-取代苯乙烯中的应用(如图7)。通过实验结果,作者得出结论,所观察到的(E)-溴化乙烯23和RAE 22之间的串联环化/烯化的烯化步骤所产生的sp2-sp3键的形成,实际上是由自由基加成片段过程和铜催化的自由基偶联反应两步所得。此外,(Z)-溴化乙烯23E和乙烯砜23F的低收率的原因可能是单一的加成片段机制的引起的结果。
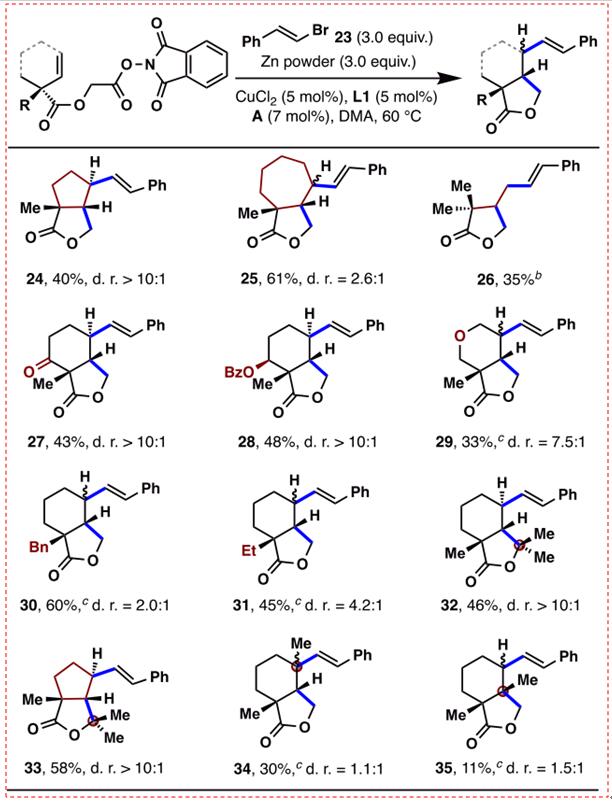
图8:环化片段的应用范围(图片来源于JACS期刊)
研究者随后探索了此串联环化/烯基化反应在构建结构多样化的内酯系统的能力,实验结果如图8所示。季碳中心的构建仍然是有机合成中的一个挑战,令人欣慰的是,季碳中心可以在最初的环化位置被成功构建,得到的产物32和33被分离。此外,尽管乙烯基烯烃34的收率相对较低(30%),但也可以在烯基化位置构建季碳中心;而在35的季碳中心构建时,结果并不理想。
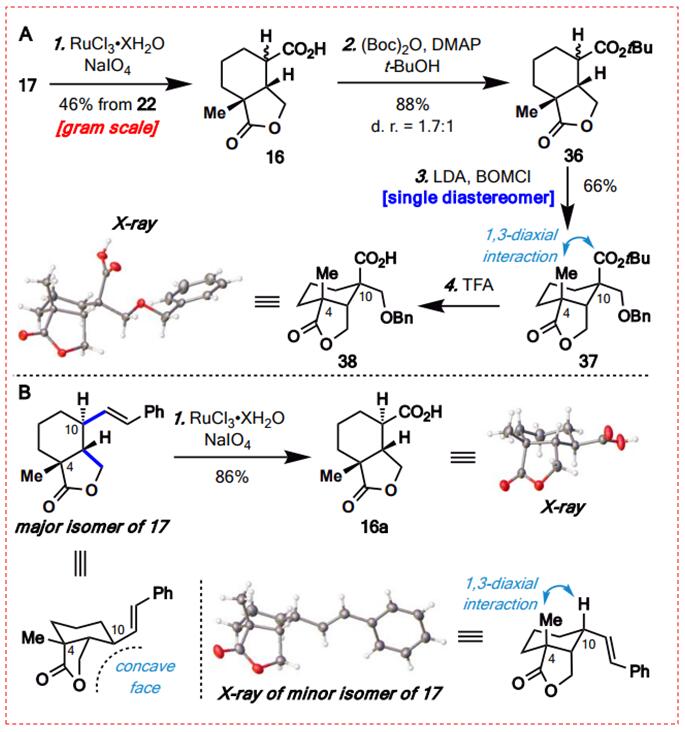
图9:C10季碳立体中心的构建(图片来源于JACS期刊)
通过RuCl3和NaO4氧化分解苯乙烯17,从氧化还原酯(RAE)22中以46%的产率获得羧酸16。利用Takeda方法将16与Boc2o酯化,得到88%产率的酯36,作为1.7:1的非对映体混合物。有趣的是,将36的混合非对映体与低密度脂蛋白(LDA)处理,然后添加Bomcl,得到37个单一的非对映体。研究人员推测在新形成的季碳中心观察到的高立体选择性应归因于C4轴向甲基产生的1,3-二轴相互作用。因此,苄基甲基醚部分被安装在较小的位阻面上。通过对38的X射线晶体学分析进一步证实了37的结构,该分析由37的TBU酯在TFA存在下的脱保护提供。
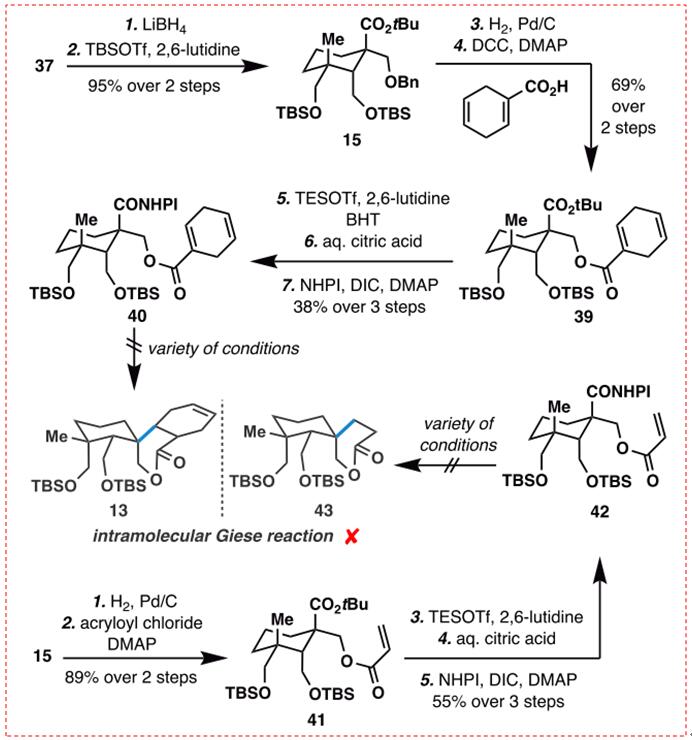
图10:分子内Giese反应的初步研究(图片来源于JACS期刊)
从逻辑上讲,根据1,3-二轴相互作用对C10季碳中心立体化学结果影响,在酸38上的直接脱羧的Giese反应可能会产生不需要的立体异构体。为了克服上述凹面效应的1,3-二轴相互作用,37中的内酯部分被LiBH4降低,得到的二醇是由两个大的TBS基团保护的。苄基醚15加氢后,新形成的醇与环己烷1,4-二烯-1-甲酸酯化得到酯39,收率69%。在对反应条件进行了广泛筛选后,研究者发现在BHT存在下用Tesotf和2,6-lutine处理39,然后用AQ水解得到的TES酯。柠檬酸产生所需的羧酸,25与NHPI(NHPI)顺序活化,以中等产量给出RAE 40。不幸的是,提出的分子内脱羧基Giese反应被证明是有问题的。由于Rae 40受到Baran还原性脱羧条件或Overman可见光光光催化条件的影响,产生了一种复杂的混合物。10c-e这些失败主要是由于直接脱羧、烯烃迁移以及基于粗核磁共振分析的其他未知副反应造成的。用麦克米伦的光催化条件直接处理RAE40酸前体也没有结果。
为了避免1,4-环己二烯部分的不稳定性,研究者接下来使用一种简单的丙烯酸酯来进行6-内三环化反应。通过5个步以良好的收率从化合物15开始顺利地制备了丙烯酸酯42。然而,RAE 42暴露在Baran的还原性脱羧条件下导致了RAE的直接脱羧和水解:未观察到所需的内酯43。提高反应温度只会导致分解。将RAE 42或其前体酸暴露于各种光催化条件下同样不成功。10b-d作者推测,高张力的螺-内酯过渡状态和左手边拥挤的环己烷环可能阻止了环化过程。
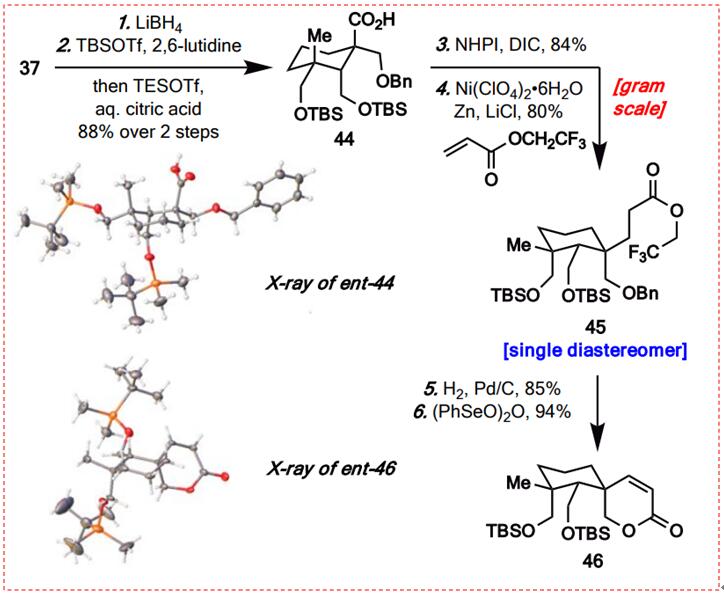
图11:分子间Giese反应激发的螺内酯的合成(图片来源于JACS期刊)
随后,研究者通过LiBH4将内酯部分还原为37,随后进行一锅反应以保护生成的二醇和TBS基团,并用Tesotf脱除TButyl酯保护,得到88%产率的酸44。X-射线晶体学分析证实了酸性44的结构,表明环己烷的底面被一个大的TBS基团阻挡,从而为随后从顶面开始的脱羧Giese反应提供了合适的途径。为此,在THF中使用N-羟基邻苯二甲酰亚胺(NHPI)和DIC直接活化44可提供84%产率的相应RAE;使用DMAP或CH2Cl2可导致较低产率。将所得的RAE暴露于含镍(ClO4)2和锌的2,2,2-三氟甲基丙烯酸酯中,成功地提供了所需的45作为单一的非对映体,产率为80%。10E在氢化条件下去除苄基,然后自发内酯化,生成关键的螺旋内酯,这是由于用苯二酸酐氧化CH,得到高产率的不饱和内酯46,并通过X射线晶体学分析证实了46的结构(合成路线如图11所示)。
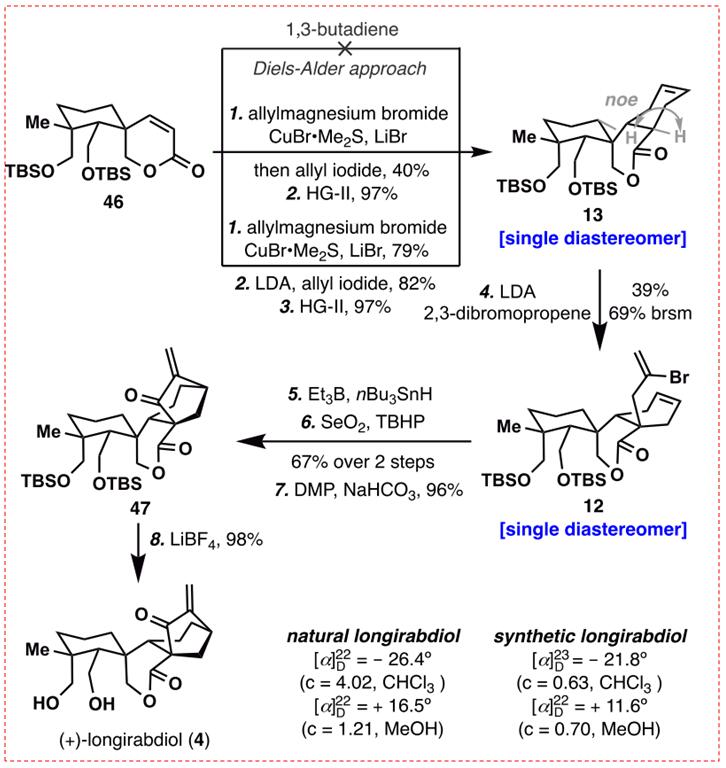
图12:Longirabdiol (4)的合成(图片来源于JACS期刊)
在获得螺内酯46后,研究人员下一步探索了桥接环系统来完成全部的合成。事实证明,通过二烯烃46和1,3-丁二烯之间的Diels-Alder反应合成13似乎很简单,在对各种参数(温度、压力、加入或不加入路易斯酸添加剂)进行条件筛选,但没有拿到目标产物。此步反应。作者采用了两个步进行:将烯丙基铜酸酯类立体选择性共轭添加到46,然后用碘化烯丙基处理,得到一个中等产率的二烯,作为一个单独的非对映体。随后,该二烯的RCM反应以97%的产率提供所需的化合物13。通过核磁共振来确证了13的结构。用LDA脱除13,然后用2,3-二溴丙烷烯丙基化,以 39%的收率得到12作为唯一的非对映异构体(同时回收率为51%的13)。研究者曾尝试改善此反应的转化率,但是没有满意的结果。将12暴露于NBU3SNH和ET3B引发5-exo自由基消除,由此产生的双环[3.2.1]辛烷经过烯丙基氧化,经2步产生67%的醇。新形成的醇的经Dess-Martin氧化,然后使用LiBF4进行脱保护,给出longrabdool(4),经波普鉴定确证了4的结构(图12)。研究者随后用longrabdool(4)作为原料,顺利合成了5和6天然产物。
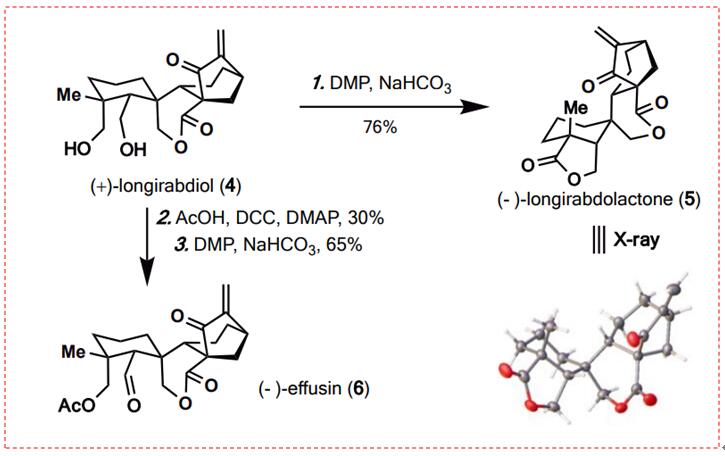
图13:longirabdolactone (5)和effusin (6)的合成(图片来源于JACS期刊)
从伊索唐属植物中提取的粗提取物已被证明具有抗肿瘤作用,已知的细胞毒性作用来自于分离的天冬氨酸。然而,迄今为止,尚不报道longrab二醇(4)、longrabdolactone(5)和effusin(6)的特异性生物活性。在合成了这三种天然产物之后,作者研究了它们对来自7种不同人体组织的10种癌细胞株的细胞活力的活性影响。如图14所示,这些合成化合物显示出广泛且高的抗癌活性(在μm范围内);特别是,longrabdolactone(5)通常比其他两种更有效,其中A673细胞中的IC50低于1μm。此外,作为这三种化合物的IC50值在10个被检测的癌细胞系中高度相关,推测它们可能作用于相同的细胞靶点。
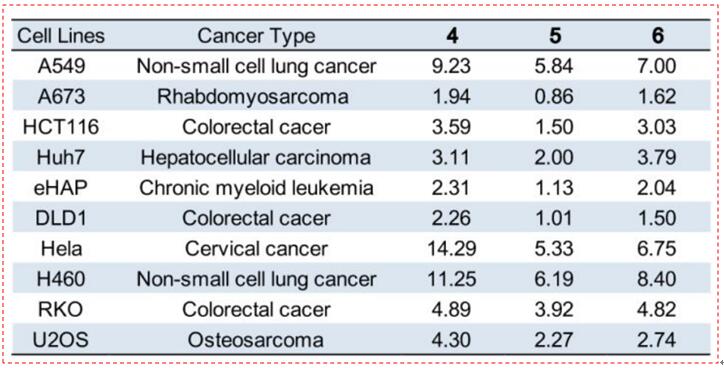
图14:合成天然产物对10种不同癌细胞株的IC50值测试(μm)(图片来源于JACS期刊)

李超研究员(图片来源于北京生命科学研究所官网)
个人履历:
2008 青岛科技大学制药工程学士
2013 北京生命科学研究所与天津大学联合培养有机合成化学博士
2014-2017年 美国斯克里普斯研究所博士后
2017-Present 北京生命科学研究所 研究员
研究方向:
(1)利用新型催化体系研究新颖的化学键构建方法,为化学分子的合成提供更理想的策略。
(2)选择与重大疾病相关的天然产物进行合成及活性优化,进而推动创新药物的研究;
(3)探索活性天然小分子的生物学作用机制和其作用靶标;
(4)设计合成能够选择性标记,追踪生物大分子或识别其相互作用的化学工具分子。
代表论文:
1. Li, C.#; Kawamata, Y.#; Nakamura, H.; Vantourout, J. C.; Liu, Z.; Hou, Q.; Bao, D.; Star, J. T.; Chen, J.; Yan, M.; Baran, P. S.* “Electrochemically Enabled, Ni-Catalyzed Amination” Angew. Chem. Int. Ed. 2017, in press
2. Li, C.#; Wang, J.#; Barton, L. M.; Yu, S.; Tian, M.; Peters, D. S.; Kumar, M.; Yu, A. W.; Johnson, K. A.; Chatterjee, A. K.; Yan, M.; Baran, P. S.* “Decarboxylative Borylation” Science, 2017, 356, 1045.
3. Qin, T.#; Cornella, J.#; Li, C.#; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell, B. D.; Eastgate, M. D.; Baran, P. S.* “A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents” Science 2016, 352, 801-805
4. Li, C.; Jones, A. X.; Lei, X.* “Synthesis and Mode of Action of Oligomeric Sesquiterpene Lactones” Nat. Prod. Rep. 2016, 33, 602-611.
5. Li, D.#; Li, C.#; Li, L.; Chen, S.; Wang, L.; Li, Q.; Wang, X.; Lei, X.*; Shen, Z.* “Natural Product Kongensin A is a Non-Canonical HSP90 Inhibitor that Blocks RIP3-dependent Necroptosis” Cell Chemical Biology 2016, 23, 257-266.
6. Hong, B.#; Li, C.#; Wang, Z.; Chen, J.; Li, H.*; Lei, X.* “Enantioselective Total Synthesis of (−)-Incarviatone A” J. Am. Chem. Soc. 2015, 137, 11946-11949.
7. Dao, H. T.; Li, C.; Michaudel, Q.; Maxwell, B. D.; Baran, P. S.* “Hydromethylation of Unactivated Olefins” J. Am. Chem. Soc. 2015, 137, 8046-8049.
8. Dong, T. #; Li, C. #; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; He, S.;* Lei, X.* “Ainsliadimer A Selectively Inhibits IKKα/β by Covalently Binding a Conserved Cysteine” Nature Commun. 2015, 6, 6522.
9. Li, C.#; Dong, T.#; Li, Q.; Lei, X.* “Probing the Anticancer Mechanism of (−)-Ainsliatrimer A through Diverted Total Synthesis and Bioorthogonal Ligation” Angew. Chem. Int. Ed. 2014, 53, 12111-12115.
10. Li, C.; Lei, X.* “Strategies toward the Biomimetic Syntheses of Oligomeric Sesquiterpenoids” J. Org. Chem. 2014, 79, 3289-3295.
11. Li, C.; Dong, T.; Dian, L.; Zhang, W.; and Lei, X.* “Biomimetic Syntheses and Structural Elucidation of the Apoptosis-Inducing Sesquiterpenoid Trimers: (-)-Ainsliatrimers A and B” Chem. Sci. 2013,4, 1163-1167. 12. Li, C.; Li, X.; Wang, X.; Lei, X.* “Diversity-oriented Synthesis of Bicyclic Ring Systems via a Conjugate Addition/aldol/RCM Process” Sci. China. Chem. 2013, 56, 337-341. 13. Li, C.; Dian, L.; Zhang, W.; Lei, X.* “Biomimetic Syntheses of (-)-Gochnatiolides A-C and (-)-Ainsliadimer B” J. Am. Chem. Soc. 2012, 134, 12414-12417. 14. Li, C.; Tu, S.; Wen, S.; Li, S.; Chang, J.; Shao, F, Lei, X.* “Total Synthesis of the G2/M DNA Damage Checkpoint Inhibitor Psilostachyin C” J. Org. Chem. 2011, 76, 3566-3570. 15. Li, C.; Yu, X.; Lei, X.* “A Biomimetic Total Synthesis of (+)-Ainsliadimer A” Org. Lett. 2010, 12, 4284-4287.