| 导 读
2019年7月15日, 浙江大学药学院胡永洲组、翁勤洁组在Journal of Medicinal Chemistry杂志(IF: 6.259),发表题为“Discovery of 3,4,6-TrisubstitutedPiperidine Derivatives as Orally Active, Low hERG Blocking Akt Inhibitors viaConformational Restriction and Structure-based Design”的论文,研究人员通过构象限制和基于结构设计的策略,发现了一个具有良好类药性的Akt抑制剂E22,该候选药具有良好的药代动力学特征,在体内外均表现出超强的抗肿瘤作用,通过机制研究发现化合物E22可以显著抑制肿瘤细胞和组织中的AKT激酶的磷酸化。
| 内 容
AKT,又称为蛋白激酶B(简称PKB),是一种丝氨酸/苏氨酸激酶,属于AGC激酶家族,与PKA和PKC具有高度的同源性。AKT可分为三种类型的同工酶,分别是AKT1、AKT2和AKT3,这三种同工酶之间又具有高度的同源性和相似的下游靶点。因此,这对研发高效低毒的AKT抑制剂带来了巨大的挑战。
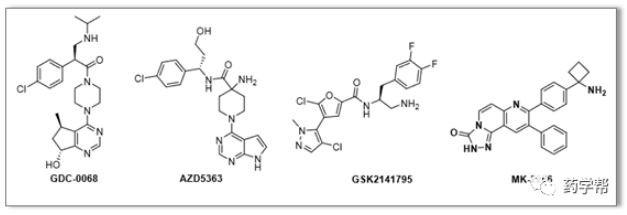
图1. 典型的AKT抑制剂的化学结构(图片来源于JMC期刊)
目前,处于开发阶段的AKT抑制剂,包括GDC0068,AZD5363,GSK2141795和MK2206等代表性的候选药。其中英派药业的GDC0068已完成II期临床试验,正在进行III临床实验,申报的适应症包括三阴性乳腺癌和转移性前列腺癌的治疗。此外,AZD5363(Capivasertib)与紫杉醇联合治疗三阴性乳腺癌的临床研究结果显示,可显著延长患者的生存期。另外,GSK2141795(也称为GSK-795或Uprosertib)是葛兰素史克公司开发的另一种具有ATP竞争性的AKT抑制剂,目前正在早期临床试验中评估其对不同类型癌症的治疗效果,如多发性骨髓瘤、乳腺癌等。这些临床阶段的候选药物各俱优点,如AZD5363的HERG阻断率低,GDC0068的选择性高,GSK-795的有效剂量低,PK分布良好,这些候选物为寻找新的药物提供了重要依据。
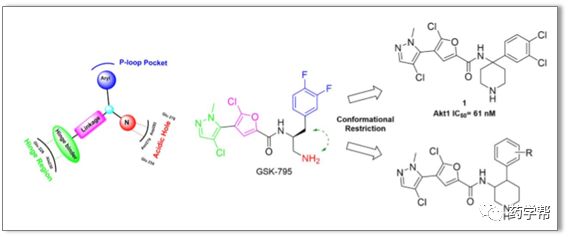
图2. 基于GSK-795结构的设计方法(图片来源于JMC期刊)
近日,浙江大学药学院胡永洲组、翁勤洁组在AKT抑制剂的研发领域取得了重大突破。研究人员巧妙的通过构象限制和基于结构设计的策略,获得了一系列3,4-二取代哌啶衍生物,并系统地研究了先导化合物A12的构效关系,包括苯基、铰链连接片段和哌啶部分的取代修饰,最终发现了一个良好类药性的3,4,6-三取代哌啶衍生物E22。
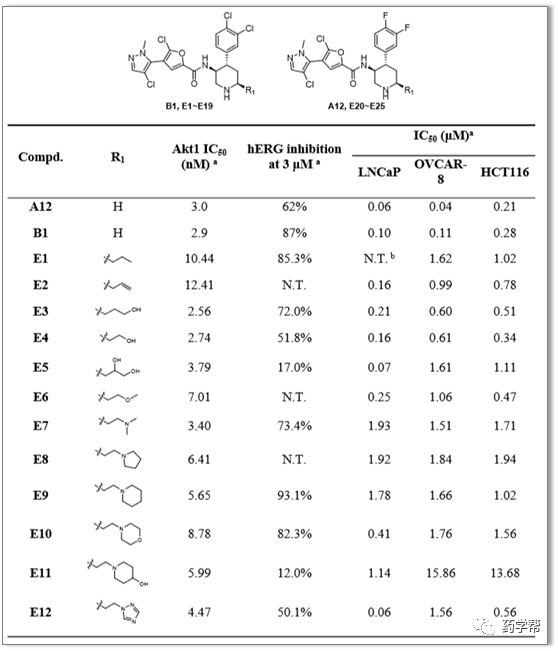
图3. 化合物E1-E12的AKT1酶活性和癌细胞系活性(图片来源于JMC期刊)
前期,研究者通过对3,4二取代哌啶衍生物的SAR研究,发现AKT和HERG的抑制活性不能很好地平衡。可能由于哌啶的5位或6位朝向溶剂区,因此这是提供总体平衡的关键,特别是关于AKT抑制作用。在合成两种骨架的(3S,4S,6S)-三取代哌啶衍生物中,一系列引入的侧链都可以很好地耐受。另外,侧链的长度对化合物的AKT1抑制活性影响较小,大部分化合物的IC50值均在10nm以下。具有亲水侧链的化合物在AKT1抑制方面表现更好。如图4, 5。
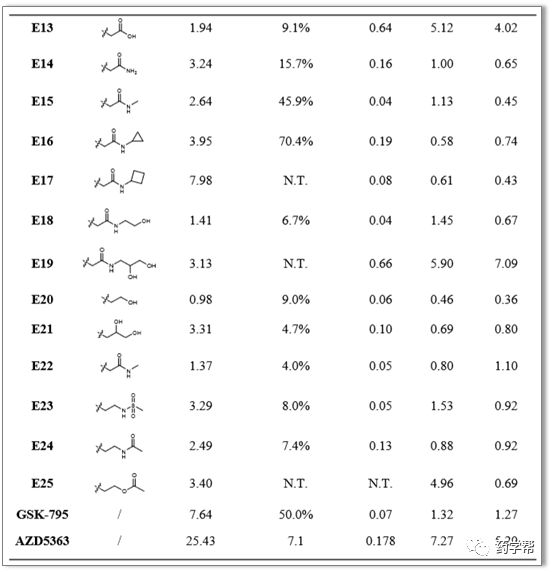
图4. 化合物E13-E25的AKT1酶活性和癌细胞系活性(图片来源于JMC期刊)
根据构效关系研究结果,最优的化合物E20和E22进一步测试其PK分布。如图5所示,E22按体重剂量40 mg/kg给小鼠、大鼠口服(化合物溶解在盐水中),作者发现小鼠体内E22的吸收比大鼠好,但在小鼠体内更容易被清除。此外,E22在小鼠体内的生物利用度为48.4%,血浆蛋白结合率高(93%),表明E22的停留时间较长。
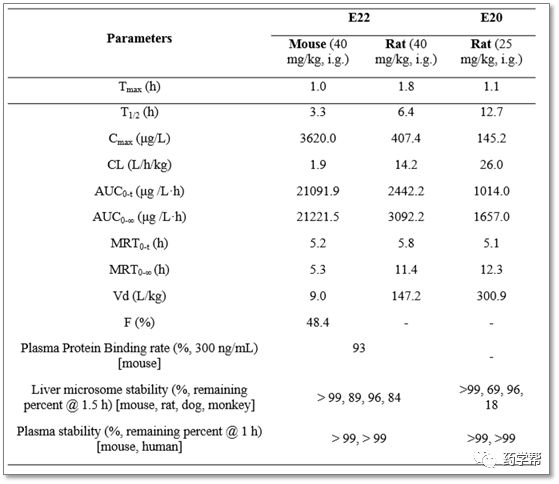
图5. 化合物E20和E22的药动学性质(图片来源于JMC期刊)
化合物E20的剂量为25 mg/kg,药代动力学实验结果显示,E20对大鼠有较好的吸收作用,且具有较好的清除率。此外,研究者还对E20和E22在肝微粒体和血浆中的体外稳定性进行了评价。这两种化合物在血浆中的稳定性较好,而化合物E20在大鼠和猴微粒体中的稳定性低于E22。
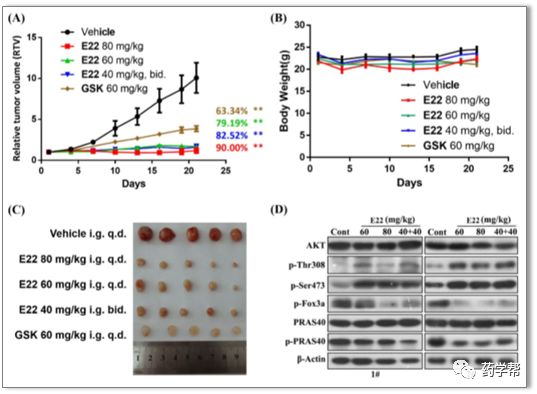
图6. 化合物E22通过抑制AKT的激酶活性来发挥抗肿瘤作用(图片来源于JMC期刊)。(a、b和c)将移植有skov3人异种移植物小鼠随机分为5组(n=5只小鼠/组),并以不同剂量(60、40+40或80 mg/kg,i.g.)、GSK-795(60 mg/kg,i.g.)或载体每天注射E22,持续21天。处死小鼠后,测定其相对肿瘤体积、肿瘤重量、肿瘤生长抑制和T/C值。数据代表平均值±标准差。(d)肿瘤组织中总Akt、P-Akt、Pras40、P-Pras40和P-Fox3a的蛋白质印迹分析。
作者在进行体内药效学实验之前,先对E22的安全性进行了评估。实验结果显示,小鼠血液生化指标一切正常。在裸鼠SKOV3卵巢癌异种移植模型中,给E22后观察到显著抑制肿瘤的生长。此外,小鼠的体重没有受到影响,且在不同剂量的治疗期间没有小鼠死亡(图6b)。在连续7天以50mg/kg/d给药后,肿瘤中可检测到560 ng/mL的E22,提示E22能有效地渗透到肿瘤组织中。体内试验完成后,研究者对肿瘤组织的进行了Western blot分析,实验表明E22对包括Fox3a和Pras40在内的AKT底物磷酸化水平具有剂量依赖性抑制作用(图6c和d)。
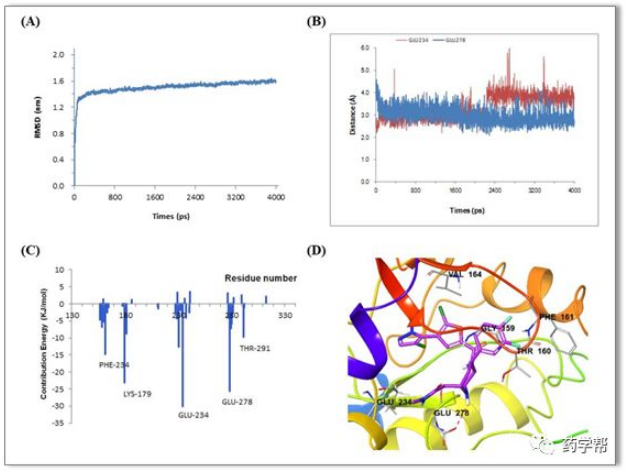
图7.化合物E22与AKT1蛋白(PDB ID: 4GV1)相互作用的MD模拟与分析(图片来源于JMC期刊)
为探索化合物E22与AKT蛋白可能的结合方式,进行了计算对接和分子动力学研究。分子对接显示E22与AKT残基Glu 278的羧基氢键结合。此外,哌啶上的6位侧链延伸到溶剂区,而4-苯基在疏水区,与gly159、thr160、phe161和val164残基相互作用(图7)。
总结:研究者通过引入一系列不同的连接基团或侧链,进行全面的构效关系研究,最终得到一个良好类药性化合物E22。这个化合物抑制所有的AKT亚型,其活性小于2 nM。同时对其他激酶,特别是非AGC家族激酶具有很好的选择性,且具有低的HERG离子通道的抑制性、良好的药代动力学特性和高的异种移植瘤抑制活性,实验结果表明E22是一个很有前景 AKT抑制剂类抗肿瘤候选药物。
附合成路线:
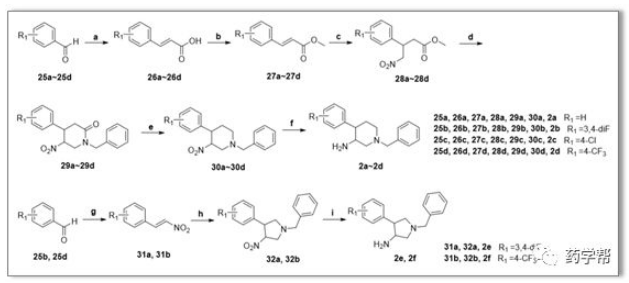
Scheme 1.(a)丙二酸、哌啶、吡啶;(b)硫酸二甲酯、K2CO3、丙酮;(c)硝基甲烷、DBU、MECN;(d)多聚甲醛、苄胺、乙氧基;(e)硼烷-甲基硫化物配合物和THF;(f)锌、ACOH、乙氧基;(g)硝基甲烷、乙酸铵、ACOH;(h)CF3COOH、N-(甲氧基甲基)-N(三甲基硅烷基甲基)苄胺类,DCM;(i)SnCl2,EtOAc
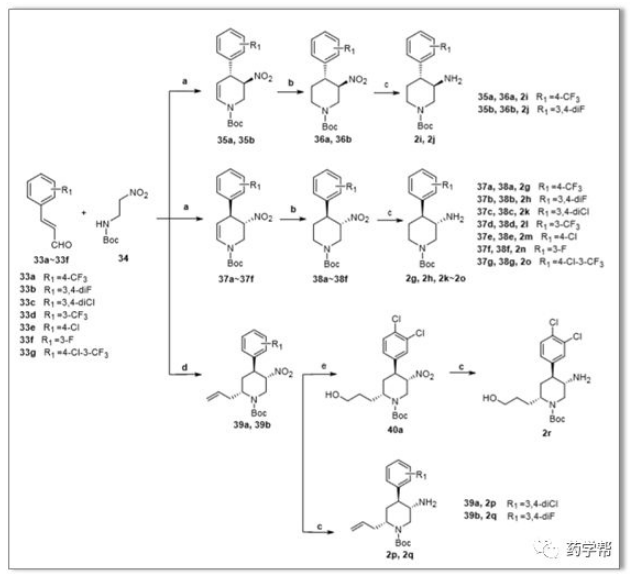
Scheme 2. 胺片段的合成路线。a)i. (S)或(R)-2-(二苯基(三甲基硅氧基)甲基)吡咯烷,二氯甲烷;ii.TFA,二氯甲烷;b)i. Et3SiH,TFA;ii. BOC2O,二氯甲烷;c)铁,NH4Cl,EtOH/H2O;d)i. (S)-2(二苯基(三甲基硅氧基)甲基)吡咯烷,DCM;ii.烯丙基三甲基硅烷,三氟化硼醚酸酯,DCM;e)硼烷-甲基硫化物络合物, NaOH,H2O2。
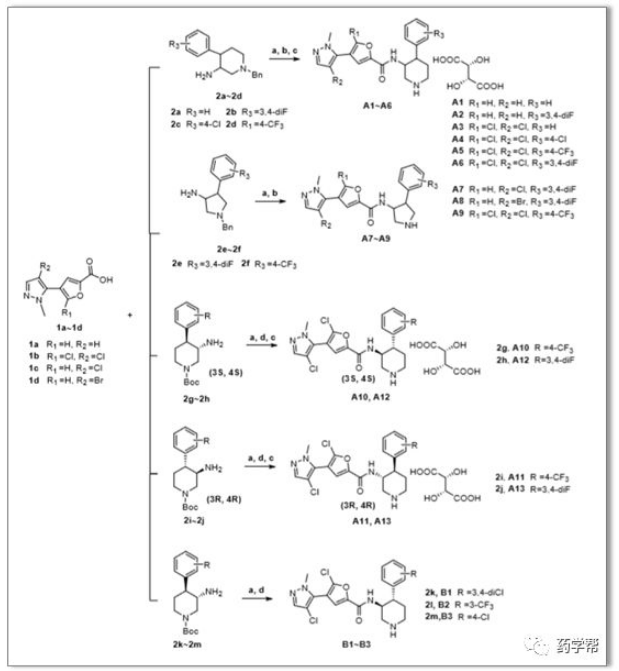
Scheme 3 .目标化合物的合成路线。a)EDCI、HOBT、DIPEA、DCM;b)氯甲酸乙酯、1,2-二氯乙烷;c)酒石酸盐;d)HCl/EA。
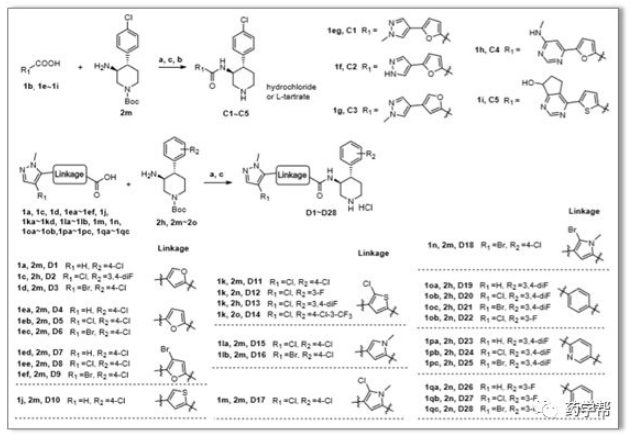
seline;"> Scheme 4.目标化合物的合成路线。a)EDCI、HOBT、DIPEA、DCM;b)酒石酸盐;c)HCl/EA。
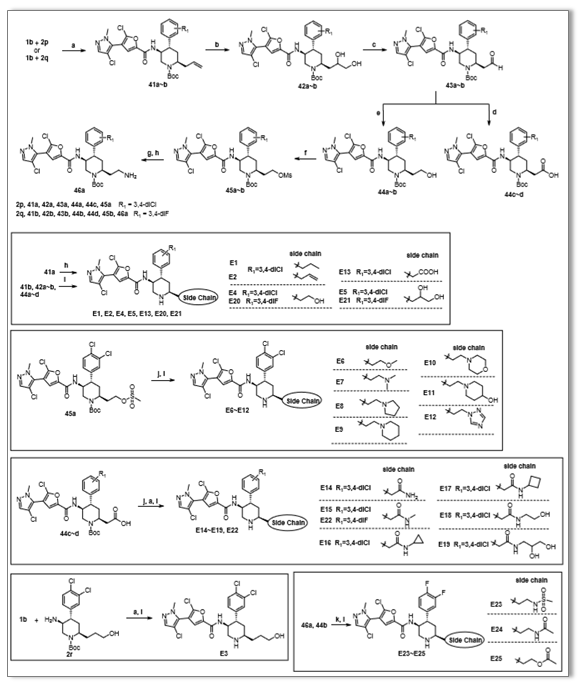
Scheme 5.目标化合物的合成路线。a)EDCI、HOBT、DIPEA、DCM;b)OSO4、NMO、THF/H2O;c)NAIO4、THF/H2O;d)NaClO2、KH2PO4、2-甲基-2-烯;e)NABH4、THF/MeOH;f)MSCL、DIPEA、DCM;g)NAN3、DCM;h)PD/C、H2、ETOAC;i)HCL/EA;j)胺或CH3ONA;k)MSCL或ACL、DIPEA。
✔胡永洲教授简介

胡永洲教授(图片来源于浙江大学官网)
| 教育工作经历
1974.9~1977.7 浙江医科大学药学专业 学生
1977.9~1987.5 浙江医科大学药学系 助教、讲师
1987.6~1987.12 日本歧阜药科大学 合作研究
1988.1~1991.7 浙江医科大学药学系 讲师
1991.8~1994.7 中科院上海药物研究所 获博士学位
1994.8~1995.5 浙江医科大学药学系 副教授
1995.6~1996.12 加拿大阿尔伯塔大学 博士后
1997.1~至今 浙江大学药学院 副教授、教授
| 研究领域
1.药物设计、合成与构效关系研究;
2.抗肿瘤、老年退行性疾病治疗药物研究;
3.生物活性糖类物质和杂环化合物合成方法学研究。
✔翁勤洁教授简介

翁勤洁教授(图片来源于浙江大学官网)
| 学历背景
2006年9月至2011年6月,浙江大学药学院,医学博士。
2002年10月至2006年6月,温州医学院,理学学士。
| 国外经历
2008年12月至2010年12月,美国University of Texas Southwestern Medical Center at Dallas联合培养。
| 工作经历
2011年7月-2013年4月 浙江大学药学院,博士后,助理研究员
2013年4月-至今,浙江大学药学院,助理研究员、副教授、GLP中心副主任
2015年3月-2016年5月,国家食品药品监督管理总局挂职
| 研究方向
1)少突胶质细胞发育调控及相关疾病模型机制研究
2)药物免疫毒理学机制研究
3)创新药物研发
@ 该文同时在药学帮公众号发布,欢迎关注公众号查看。