| 导 读
2019年7月16日, 暨南大学丁克课题组、陆小云课题组在Journal of Medicinal Chemistry (IF: 6.259) 杂志上,发表题为“2-Amino-2, 3-Dihydro-1H-Indene-5- Carboxamide-basedDiscoidin Domain Receptors 1 (DDR1) Inhibitors: Design, Synthesis, and In VivoAnti-pancreatic Cancer Efficacy”的研究论文,研究人员发现了一个新型高选择性DDR1抑制剂,IC50达14.9 nM,且该化合物具有良好的药代动力学特性和体内治疗效果。
原文地址:https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00365
| 内 容
胰腺癌,又被称为“癌症之王”,5年生存率低于7%,临床一线药物如吉西他滨容易产生化疗耐药性,目前缺乏针对胰腺癌有效的治疗方法,一般治疗预后效果较差。
研究表明,胰腺癌患者化疗耐药的关键因素是去纤维增生和肿瘤微环境。其中结皮增生是胰腺癌的一个显著病理特征,是由于癌相关成纤维细胞迅速扩张和细胞外基质成分沉积所致。大量研究表明,细胞外基质与胰腺癌的发展、免疫逃避和治疗抵抗是密切相关的。因此,基质靶向治疗成为一种备受关注的提高治疗反应的策略,胶原是细胞外基质中最丰富的成分,胶原的生物学功能主要由整合素和盘状结构域受体(DDR)介导, DDR是一类细胞外基质,属于受体酪氨酸激酶RTK家族。
迄今为止已鉴定出两个DDR成员(DDR1和DDR2)。DDR1主要在上皮细胞中表达,而DDR2通常存在于结缔组织的细胞中。DDR1的失调经常在多种人类癌症中被发现,并参与一些关键的细胞过程,如细胞分化、增殖、粘附、迁移和侵袭。研究表明,DDR1的过度表达介导了B细胞的生存前信号和转移,并参与某些类型癌症的复发。DDR1也被证明有助于胰腺癌上皮间充质转换。
通过选择性DDR1抑制剂7rh对DDR1的药理学抑制,成功减缓了肿瘤的进展,增强了对标准治疗胰腺癌方案的化疗敏感性。DDR1被认为是抗胰腺癌药物发现的新分子靶点。
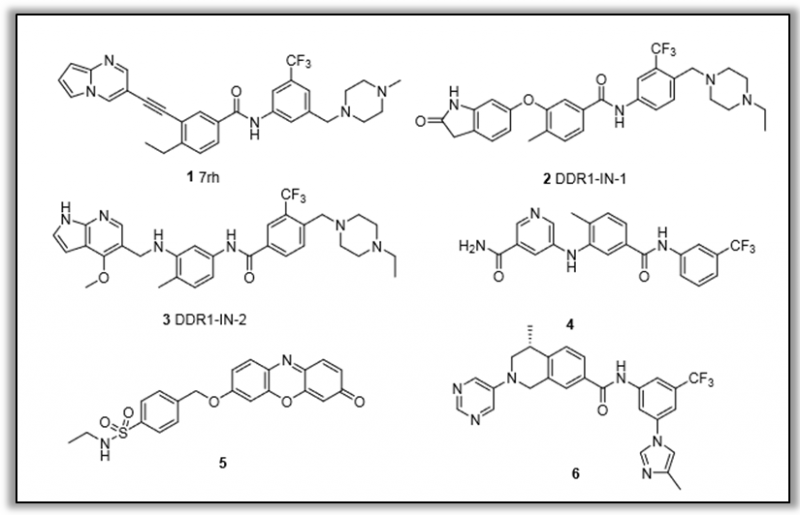
图1.报道的选择性DDRs抑制剂(图片来源于JMC期刊)
暨南大学丁克课题组、陆小云课题组,继7月12日,发表到JMC杂志关于EGFR三重突变体抑制剂的研究之后,同月内时隔4日于7月16日又一精彩研究再次刊登JMC期刊。此次研究,课题组设计合成了一系列2-氨基-2,3-二氢-1H-茚-5-酰胺类化合物作为新型选择性DDR1抑制剂。
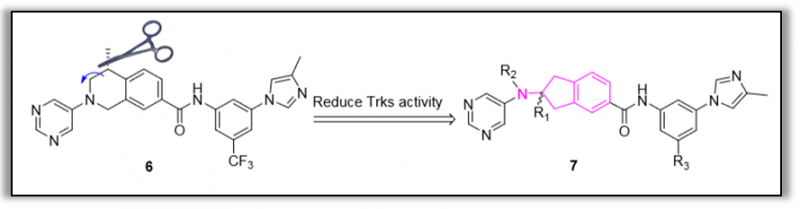
图2.2-氨基-2,3-二氢-1H-吲哚-5-甲酰胺衍生物作为新型Trks保留的选择性DDR1抑制剂的设计(图片来源于JMC期刊)
研究人员发现了一种具有代表性的化合物7f,与DDR1结合,Kd值为5.9nM,其抑制激酶活性IC50值为14.9nM。7f能有效抑制胶原诱导的DDR1信号传导和上皮间充质转化,且剂量依赖性的抑制胰腺癌细胞的集落的形成,在胰腺癌原位小鼠模型中显示出良好的体内治疗效果。
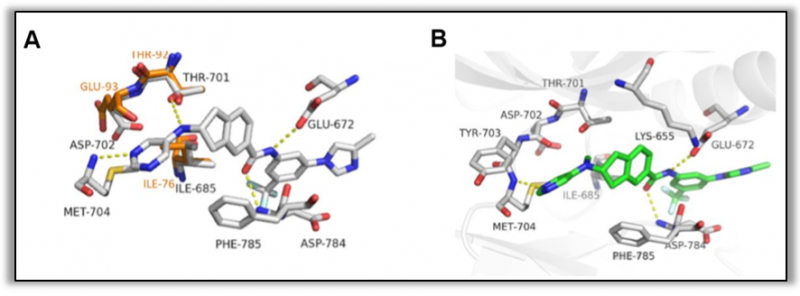
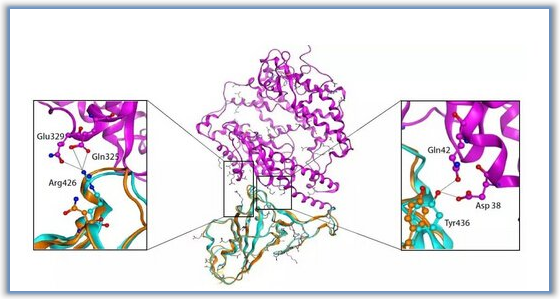
图3.A.DDR1(PDB:6HP9)和DDR2的叠加;B. 7f分子对接DDR1(PDB:6HP9)(图片来源于JMC期刊)
总结:课题组采用基于结构的药物设计方法,设计合成了一系列2-氨基-2,3-二氢-1H-茚-5-甲酰胺衍生物作为新型高选择性DDR1抑制剂。化合物7f以14.9nM的IC50值强烈抑制DDR1,而对其他403种1.0μM的非突变激酶的作用明显减弱。7f还有效抑制由DDR1诱导的胶原诱导的钙粘蛋白转换,并剂量依赖性抑制胰腺癌细胞的集落形成。此外,7f在胰腺癌原位同基因模型中口服给药的结果显示具体良好的药代动力学特性和体内治疗效果,7f作为一个潜在候选药正在进行更深入的生物学研究。
附录:化合物合成路线
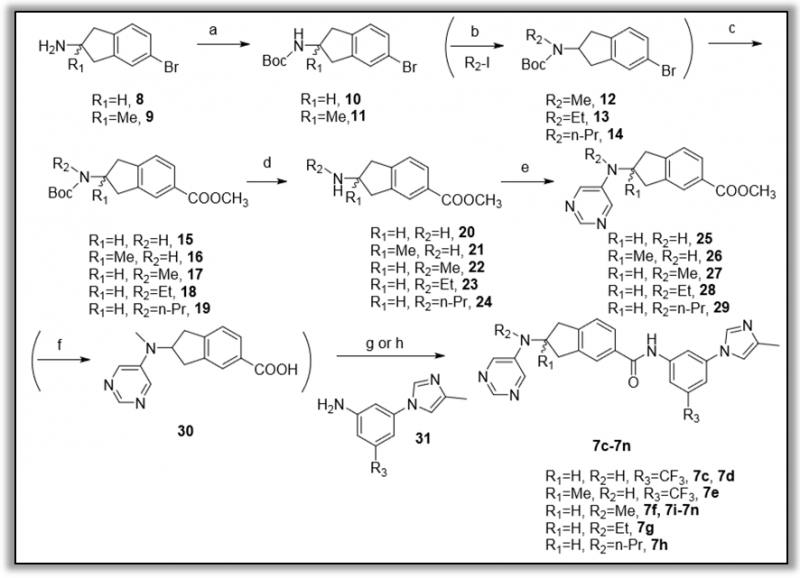
Scheme 1. 化合物合成路线(图片来源于JMC期刊)
(a)二碳酸二叔丁基酯(BOC2O),三乙胺(Et3N),90-95%;(b)氢化钠(NaH),N,N-二甲基甲酰胺(DMF),碘甲烷,12;碘乙烷,13;碘丙烷,14;50-80%; (c)钯(ii)乙酸(Pd(OAC)2),Et3N,一氧化碳(CO),100℃,50%, 4M HCl;(d)HCM; (e)亚苄叉丙酮)钯(Pd(dba)2),2-二环己基膦-2’,6’-二异丙氧基联苯(Ruphos),碳酸铯(CS2CO3),5-溴嘧啶,80℃,30-40%;(f)1M氢氧化钠(NaOH),二恶烷,98%; (g)叔丁氧基钾(t-BuOK),20℃,30%,70%; (h)1H-1H-三唑1-基)-1,1,3,3-四甲基尿嘧啶六氟磷酸(HATU),N,N-二异丙基乙胺(DIPEA),20-55%。
✔丁克教授简介

丁克教授(图片来源于暨南大学官网)
丁克,1973年12月出生。英国皇家化学会Fellow
暨南大学药学院教授、博士生导师。
【教育经历】
2001/12 ~ 2005/2 美国密西根大学(the University of Michigan,Ann Arbor)
肿瘤 中心,博士后研究。导师:Shaomeng Wang 教授。
1998/9 ~ 2001/7复旦大学,化学系(与上海有机化学研究所联合培养)
生物有机化学博士。导师:马大为教授。
1995/9 ~ 1998/7中国药科大学,药学院,药物化学硕士。导师:邢为凡教授。
1991/9 ~ 1995/7 中国药科大学,药学院,药学学士,化学制药专业。
【工作经历】
2016/6 ~ 暨南大学药学院院长、教授。
2008/10 ~ 2016/6 中国科学院广州生物医药与健康研究院, 研究员,副所长。
2008/06,入选中科院百人计划
2006/03 ~ 至今 中国科学院广州生物医药与健康研究院,研究员,博士生导师。
2005/03 ~ 2006/03美国密西根大学,医学院,Research Investigator。
【荣誉奖励】
2017,教育部“长江学者特聘教授”
2016,中组部“国家万人计划领军人才”
2016,英国皇家化学会Fellow
2015,科技部“中青年科技创新领军人才”
2014,获得国家杰出青年基金
2014,第三批“百名南粤杰出人才培养工程”支持计划
2012,国务院政府特殊津贴
2012,第六届药明康德生命化学研究奖
2012,“中科院百人计划”期终评估优秀(全中科院比例<20%)
2009,第十届“广东省丁颖科技奖”
【学术任职】
2015,中国药学会药物化学专业委员会副主任委员;
2015,中国药学会应用药理学专业委员会委员。
2015,MedChemComm. 副主编
2014,J. Chin. Pharm. Sci.(中国药学会), 编委;
2013,J. Med. Chem. 顾问编委(Advisory Editorial Board Member);
2011,中国药学会药物化学专业委员会,委员;
2010, ACS Med. Chem. Lett.,顾问编委(Advisory Editorial Board Member);
2010,广州市科学技术协会第九届委员会委员;
2009,广东省药学会药物化学专业委员会,副主任委员;
【研究领域】
重点围绕肿瘤和代谢性疾病等重大临床需求,设计和合成同时具有生物活性和成药性
的 “成(类)药性先导化合物”,为创新药物研究奠定基础。
近年来,成功地设计和合成了:
1)世界首个亚型选择性ERRa激动剂(国际同行评价)作为新型抗代谢性疾病药物候
选物(转让香港CASIGEN Pharma Inc.公司);
2)可克服慢性粒性白血病(CML)临床耐药的新一代Bcr-AblT315I突变体抑制剂(转
让广州顺健,已获CFDA临床批件);
3)可克服非小细胞肺癌(NSCLC)对Gefitinib等临床获得性耐药的全新特异性
EGFRT790M突变抑制剂(转让南京奥赛康药业);
4)具有治疗肿瘤和肺纤维化的世界首个选择性DDR1抑制剂(国际同行评价);
5)可克服耐药结核菌的新型抗结核分子(转让广州艾格医药)等。
6)早期在美国还设计和合成了Spirooxindole类p53-MDM2相互作用阻断剂转让世界
著名大型药企Sanofi,并进入I期临床研究。
✔陆小云研究员简介

陆小云研究员(图片来源于百度百科)
【个人简介】
陆小云,女,博士,暨南大学研究员,广东省杰出青年基金获得者,863青年科学家项目获得者,广东省特支计划青年拔尖人才,广州市珠江新星。2005年6月毕业于中国药科大学制药工程专业,获学士学位;2010年6月毕业于中国药科大学药物化学专业,获理学博士学位;2010年7月-2016年2月在中国科学院广州生物医药与健康研究院从事科研工作,2012年1月被聘为副研究员;2016年3月加入暨南大学药学院,从事教学科研工作。
从事创新药物先导化合物的发现研究。主要瞄准肺部疾病(肺癌和结核病等)临床耐药问题,设计和合成具有重要生物学功能和良好成药性的先导化合物,为创新药物开发奠定基础。现主持863青年科学家项目,广东省杰出青年项目,广东省特支计划青年拔尖人才和广州市珠江新星等研究课题。已在Angew. Chem. Int. Ed.和J. Med. Chem 等重要专业期刊发表第一作者或通讯作者SCI论文30余篇,申请发明专利20余项(授权10项,PCT 4项),部分专利已实现转移转化。曾获中国药学会-施维雅药物化学奖,中科院卢嘉锡青人才奖等奖项。
【研究领域】
1. 新型抗肺癌成药性先导化合物的研究
2. 克服临床耐药的新型抗结核药物研究
3. 其他新型激酶类抑制剂的设计研究